



Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond



Abstract
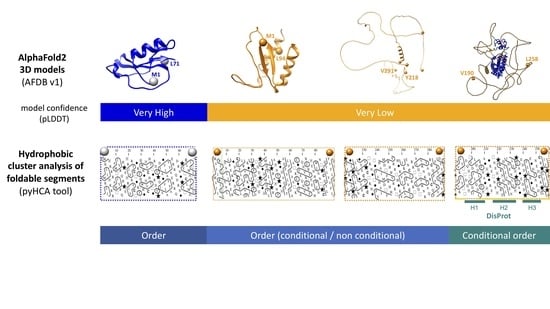
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Proteomes from AlphaFold Protein Structure Database v1
2.2. Delineation of Soluble-Like Foldable Segments within Protein Sequences
2.3. Description of Sequence and Structural Features
2.3.1. Per-Residue Disorder Prediction
2.3.2. Known Homologs
2.3.3. Secondary Structure Assignment
2.3.4. Solvent Accessibility
2.3.5. 3D Structure Comparison
2.3.6. Figure Creation
3. Results
3.1. General Features of Full-VL and Full-VH Segments from AFDB v1
3.2. Full-VH Segments
3.3. Full-VL Segments
3.3.1. Full-VL Segments with AF2 Well-folded Models
3.3.2. Full-VL Segments with AF2 Unfolded Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Akdel, M.; Pires, D.E.V.; Porta Pardo, E.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold 2 applications. bioRxiv 2021. [Google Scholar] [CrossRef]
- Alderson, T.R.; Pritišanac, I.; Moses, A.M.; Forman-Kay, J.D. Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Binder, J.L.; Berendzen, J.; Stevens, A.O.; He, Y.; Wang, J.; Dokholyan, N.V.; Oprea, T.I. AlphaFold illuminates half of the dark human proteins. Curr. Opin. Struct. Biol. 2022, 74, 102372. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Ruiz-Serra, V.; Valentini, S.; Valencia, A. The structural coverage of the human proteome before and after AlphaFold. PLoS Comput. Biol. 2022, 18, e1009818. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.-Y.; Ren, W.; Wang, J.; Kaneko, K. The Statistical Trends of Protein Evolution: A Lesson from AlphaFold Database. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wilson, C.J.; Choy, W.Y.; Karttunen, M. AlphaFold2: A Role for Disordered Protein/Region Prediction? Int. J. Mol. Sci. 2022, 23, 4591. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; CAID Predictors; DisProt Curators; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Morris, O.M.; Torpey, J.H.; Isaacson, R.L. Intrinsically disordered proteins: Modes of binding with emphasis on disordered domains. Open Biol. 2021, 11, 210222. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef]
- Csizmók, V.; Bokor, M.; Bánki, P.; Klement, E.; Medzihradszky, K.F.; Friedrich, P.; Tompa, K.; Tompa, P. Primary contact sites in intrinsically unstructured proteins: The case of calpastatin and microtubule-associated protein 2. Biochemistry 2005, 44, 3955–3964. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Stott, K. Disordered domains in chromatin-binding proteins. Essays Biochem. 2019, 63, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays 2009, 31, 328–335. [Google Scholar] [CrossRef]
- Williams, R.W.; Xue, B.; Uversky, V.N.; Dunker, A.K. Distribution and cluster analysis of predicted intrinsically disordered protein Pfam domains. Intrinsically Disord. Proteins 2013, 1, e25724. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Oldfield, C.J.; Yan, W.; Shen, B.; Dunker, A.K. Intrinsically disordered domains: Sequence ➔ disorder ➔ function relationships. Protein Sci. 2019, 28, 1652–1663. [Google Scholar] [CrossRef]
- Bitard-Feildel, T.; Callebaut, I. Exploring the dark foldable proteome by considering hydrophobic amino acids topology. Sci. Rep. 2017, 7, 41425. [Google Scholar] [CrossRef]
- Perdigão, N.; Heinrich, J.; Stolte, C.; Sabir, K.S.; Buckley, M.J.; Tabor, B.; Signal, B.; Gloss, B.S.; Hammang, C.J.; Rost, B.; et al. Unexpected features of the dark proteome. Proc. Natl. Acad. Sci. USA 2015, 112, 15898–15903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruley, A.; Bitard-Feildel, T.; Callebaut, I.; Duprat, E. A sequence-based foldability score combined with AlphaFold2 predictions to disentangle the protein order/disorder continuum. Proteins 2022. in revision. [Google Scholar] [CrossRef]
- Bitard-Feildel, T.; Lamiable, A.; Mornon, J.-P.; Callebaut, I. Order in disorder as observed by the “Hydrophobic Cluster Analysis” of protein sequences. Proteomics 2018, 18, e1800054. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Labesse, G.; Durand, P.; Poupon, A.; Canard, L.; Chomilier, J.; Henrissat, B.; Mornon, J.-P. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell Mol. Life Sci. 1997, 53, 621–645. [Google Scholar] [CrossRef]
- Eudes, R.; Le Tuan, K.; Delettré, J.; Mornon, J.-P.; Callebaut, I. A generalized analysis of hydrophobic and loop clusters within globular protein sequences. BMC Struct. Biol. 2007, 7, 2. [Google Scholar] [CrossRef]
- Lamiable, A.; Bitard-Feildel, T.; Rebehmed, J.; Quintus, F.; Schoentgen, F.; Mornon, J.P.; Callebaut, I. A topology-based investigation of protein interaction sites using Hydrophobic Cluster Analysis. Biochimie 2019, 167, 68–80. [Google Scholar] [CrossRef]
- Faure, G.; Callebaut, I. Comprehensive repertoire of foldable regions within whole genomes. PLOS Comput. Biol. 2013, 9, e1003280. [Google Scholar] [CrossRef]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Erdos, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinegger, M.; Mirdita, M.; Söding, J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 2019, 16, 603–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C. Conservation and prediction of solvent accessibility in protein families. Proteins 1994, 20, 216–226. [Google Scholar] [CrossRef]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.B.; Bernstein, D.A.; Annis, D.S.; Misenheimer, T.M.; Hannah, B.L.; Mosher, D.F.; Keck, J.L. Structure of the calcium-rich signature domain of human thrombospondin-2. Nat. Struct. Mol. Biol. 2005, 12, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wu, S.; Wang, D.; Song, X.; Yang, M.; Zhang, W.; Huang, S.; Weng, J.; Liu, Z.; Wang, W. The importance of the compact disordered state in the fuzzy interactions between intrinsically disordered proteins. Chem. Sci. 2022, 13, 2363–2377. [Google Scholar] [CrossRef]
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol. 2012, 179, 279–288. [Google Scholar] [CrossRef]
- Kim, D.-H.; Wright, A.; Han, K.-H. An NMR study on the intrinsically disordered core transactivation domain of human glucocorticoid receptor. BMB Rep. 2017, 50, 522–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørholm, A.B.; Hendus-Altenburger, R.; Bjerre, G.; Kjaergaard, M.; Pedersen, S.F.; Kragelund, B.B. The intracellular distal tail of the Na+/H+ exchanger NHE1 is intrinsically disordered: Implications for NHE1 trafficking. Biochemistry 2011, 50, 3469–3480. [Google Scholar] [CrossRef] [PubMed]
- Ostedgaard, L.S.; Baldursson, O.; Vermeer, D.W.; Welsh, M.J.; Robertson, A.D. A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. USA 2000, 97, 5657–5662. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.M.R.; Hudson, R.P.; Kanelis, V.; Choy, W.-Y.; Thibodeau, P.H.; Thomas, P.J.; Forman-Kay, J.D. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 2007, 14, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A. SCARF1: A multifaceted, yet largely understudied, scavenger receptor. Inflamm. Res. 2018, 67, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Weatheritt, R.J.; Luck, K.; Petsalaki, E.; Davey, N.E.; Gibson, T.J. The identification of short linear motif-mediated interfaces within the human interactome. Bioinformatics 2012, 28, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Lescasse, R.; Pobiega, S.; Callebaut, I.; Marcand, S. End-joining inhibition at telomeres requires the translocase and polySUMO-dependent ubiquitin ligase Uls1. EMBO J. 2013, 32, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausrath, A.C.; Kingston, R.L. Conditionally disordered proteins: Bringing the environment back into the fold. Cell Mol. Life Sci. 2017, 74, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Schnatwinkel, J.; Paissoni, C.; Andersson, E.; Herrmann, C.; Camilloni, C.; Jemth, P. Disordered Regions Flanking the Binding Interface Modulate Affinity between CBP and NCOA. J. Mol. Biol. 2022, 434, 167643. [Google Scholar] [CrossRef] [PubMed]
- Schütze, K.; Heinzinger, M.; Steinegger, M.; Rost, B. Nearest neighbor search on embeddings rapidly identifies distant protein relations. bioRxiv 2022. [Google Scholar] [CrossRef]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Rochereau, C.; Church, G.M.; Sorger, P.K.; AlQuraishi, M. Single-sequence protein structure prediction using language models from deep learning. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sen, N.; Anishchenko, I.; Bordin, N.; Sillitoe, I.; Velankar, S.; Baker, D.; Orengo, C. Characterizing and explaining the impact of disease-associated mutations in proteins without known structures or structural homologs. Brief. Bioinform. 2022, 23, bbac187. [Google Scholar] [CrossRef] [PubMed]
- Vakirlis, N.; Hebert, A.S.; Opulente, D.A.; Achaz, G.; Hittinger, C.T.; Fischer, G.; Coon, J.J.; Lafontaine, I. A Molecular Portrait of De Novo Genes in Yeasts. Mol. Biol. Evol. 2017, 35, 631–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espada, R.; Parra, R.G.; Mora, T.; Walczak, A.M.; Ferreiro, D.U. Capturing coevolutionary signals inrepeat proteins. BMC Bioinform. 2015, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heames, B.; Schmitz, J.; Bornberg-Bauer, E. A Continuum of Evolving De Novo Genes Drives Protein-Coding Novelty in Drosophila. J. Mol. Evol. 2020, 88, 382–398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Faraggi, E.; Li, Z.; Zhou, Y. Intrinsically semi-disordered state and its role in induced folding and protein aggregation. Cell Biochem. Biophys. 2013, 67, 1193–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvunis, A.R.; Rolland, T.; Wapinski, I.; Calderwood, M.A.; Yildirim, M.A.; Simonis, N.; Charloteaux, B.; Hidalgo, C.A.; Barbette, J.; Santhanam, B.; et al. Proto-genes and de novo gene birth. Nature 2012, 487, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Vakirlis, N.; Acar, O.; Hsu, B.; Castilho Coelho, N.; Van Oss, S.B.; Wacholder, A.; Medetgul-Ernar, K.; Bowman, R.W., 2nd; Hines, C.P.; Iannotta, J.; et al. De novo emergence of adaptive membrane proteins from thymine-rich genomic sequences. Nat. Commun. 2020, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.A.; Foy, S.G.; Neme, R.; Masel, J. Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nat. Ecol. Evol. 2017, 1, 0146. [Google Scholar] [CrossRef] [Green Version]
- Bitard-Feildel, T.; Heberlein, M.; Bornberg-Bauer, E.; Callebaut, I. Detection of orphan domains in Drosophila using “hydrophobic cluster analysis”. Biochimie 2015, 119, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Bungard, D.; Copple, J.S.; Yan, J.; Chhun, J.J.; Kumirov, V.K.; Foy, S.G.; Masel, J.; Wysocki, V.H.; Cordes, M.H.J. Foldability of a Natural De Novo Evolved Protein. Structure 2017, 25, 1687–1696. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruley, A.; Mornon, J.-P.; Duprat, E.; Callebaut, I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules 2022, 12, 1467. https://doi.org/10.3390/biom12101467
Bruley A, Mornon J-P, Duprat E, Callebaut I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules. 2022; 12(10):1467. https://doi.org/10.3390/biom12101467
Chicago/Turabian StyleBruley, Apolline, Jean-Paul Mornon, Elodie Duprat, and Isabelle Callebaut. 2022. "Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond" Biomolecules 12, no. 10: 1467. https://doi.org/10.3390/biom12101467
APA StyleBruley, A., Mornon, J. -P., Duprat, E., & Callebaut, I. (2022). Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules, 12(10), 1467. https://doi.org/10.3390/biom12101467