
BabyBoom: 3-Dimensional Structure-Based Ligand and Protein Interaction Prediction by Molecular Docking

Abstract
:1. Introduction
2. Materials and Methods
2.1. Orthology, Sub-Cellular Localization, and Domain Conservation
2.2. Protein-Protein and Protein-Ligand Interactions
2.3. 3-D Structure Preparation
2.4. Docking and Clustering
2.5. Molecular Dynamics Simulation
2.6. Binding Affinity (ΔG) and Dissociation Constant (Kd)
2.7. Interfacial Residues, Peptide Preparations, and Scoring
3. Results
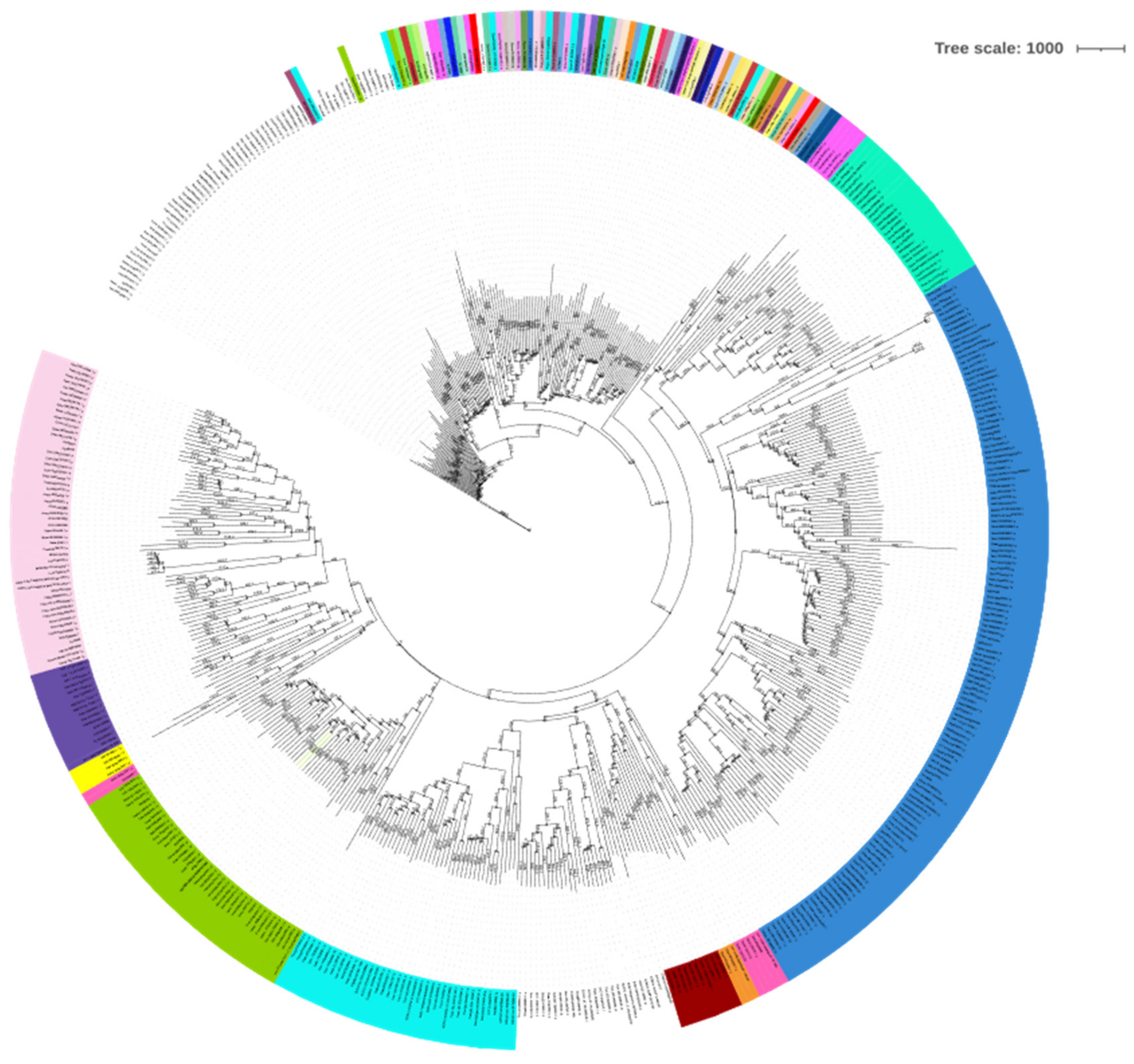
3.1. BBM Orthologs, Conservation, and Sub-Cellular Localization
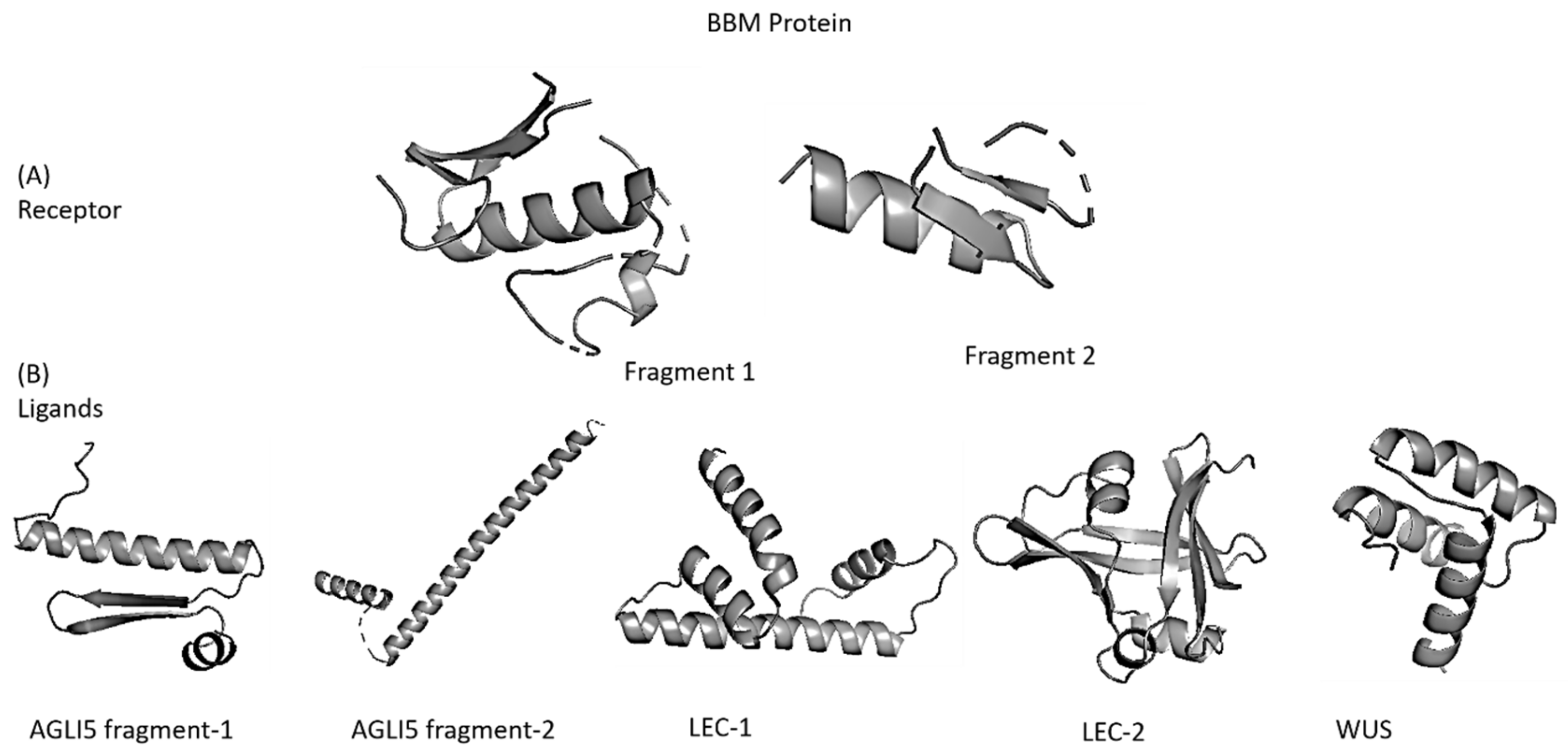
3.2. Protein-Protein and Protein-Ligand Interactions
3.2.1. Ligand Binding Site and Ligand Predictions
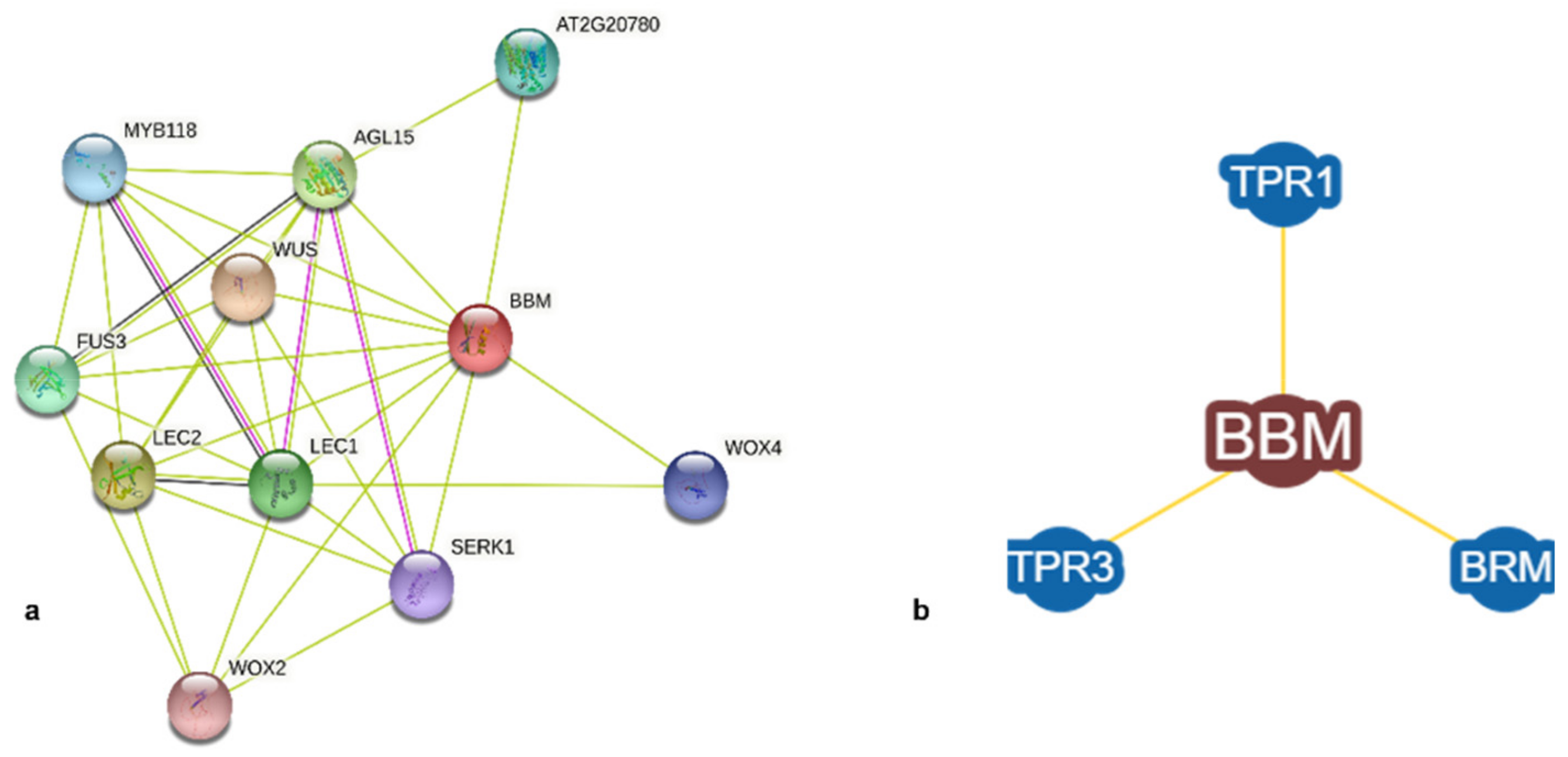
3.2.2. Sequence-Based Protein Interactions
3.3. Docking
3.3.1. Docking (BBM-Interacting Proteins) and Docked Conformation Clustering
3.3.2. Binding Energy and Dissociation Constant
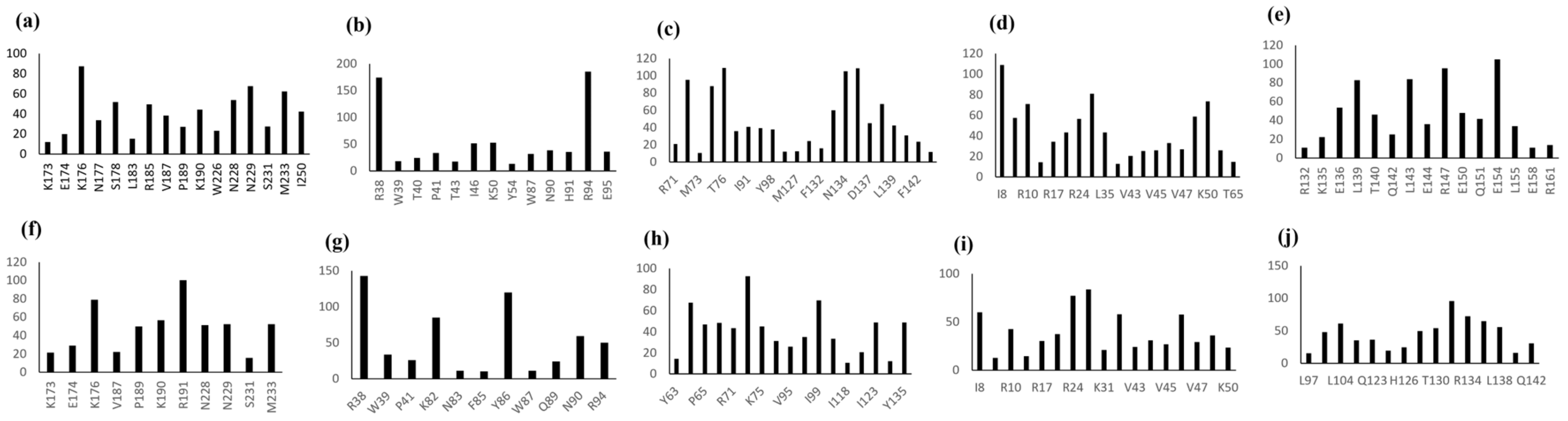
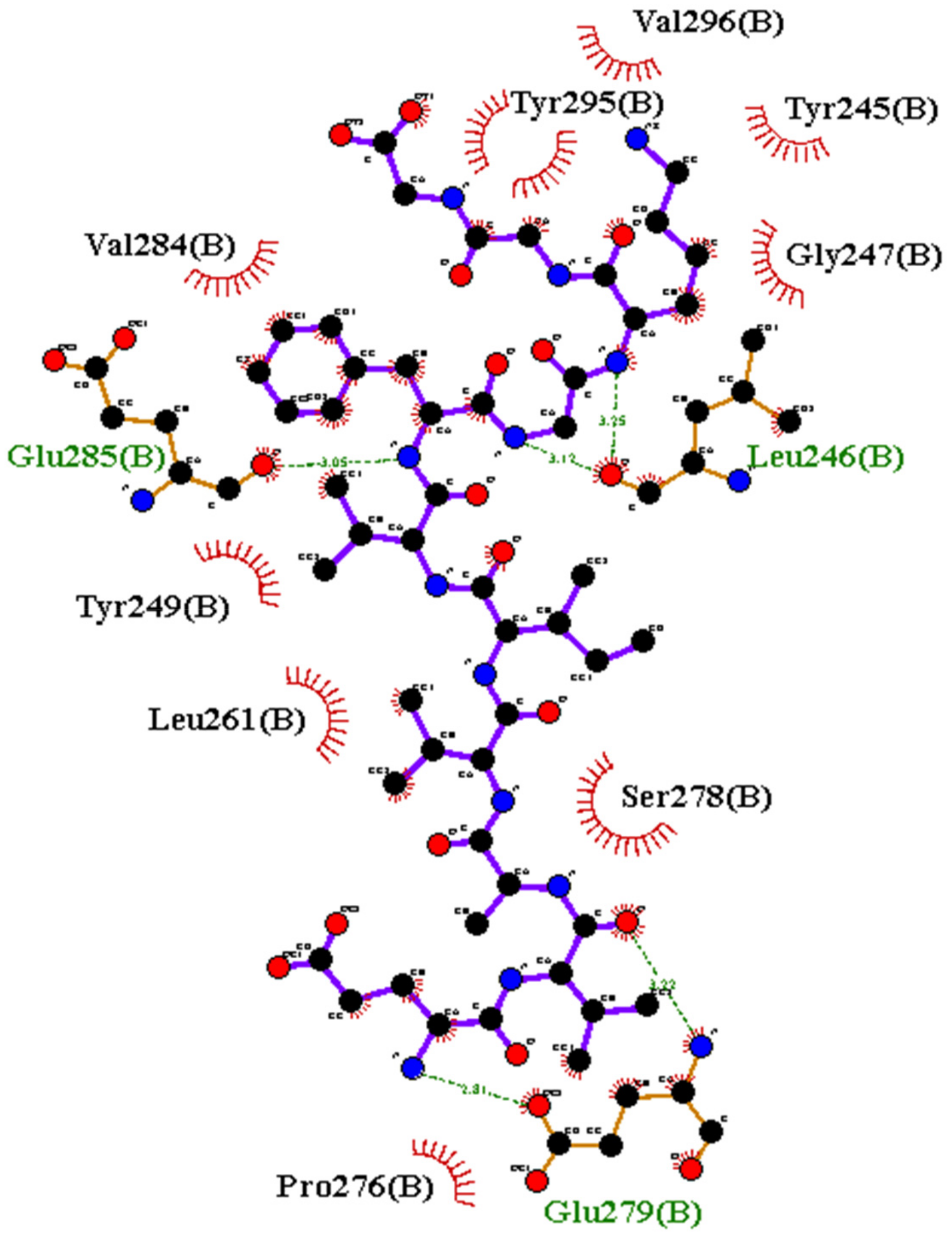
3.3.3. Interfacial Residue Stretch
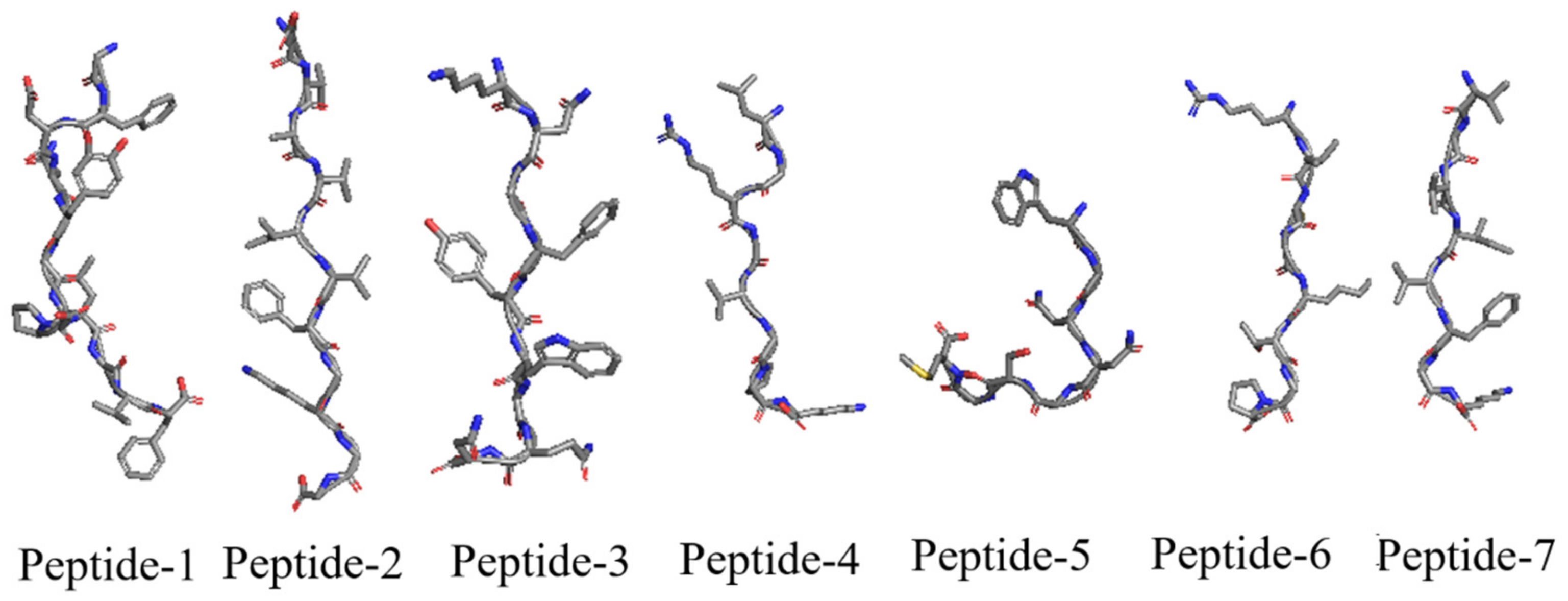
3.3.4. Peptide Preparation
3.3.5. Docking and Clustering
3.3.6. Protein-Peptide Binding Energy
3.3.7. Scoring of Peptides
3.3.8. Molecular Dynamics Simulation
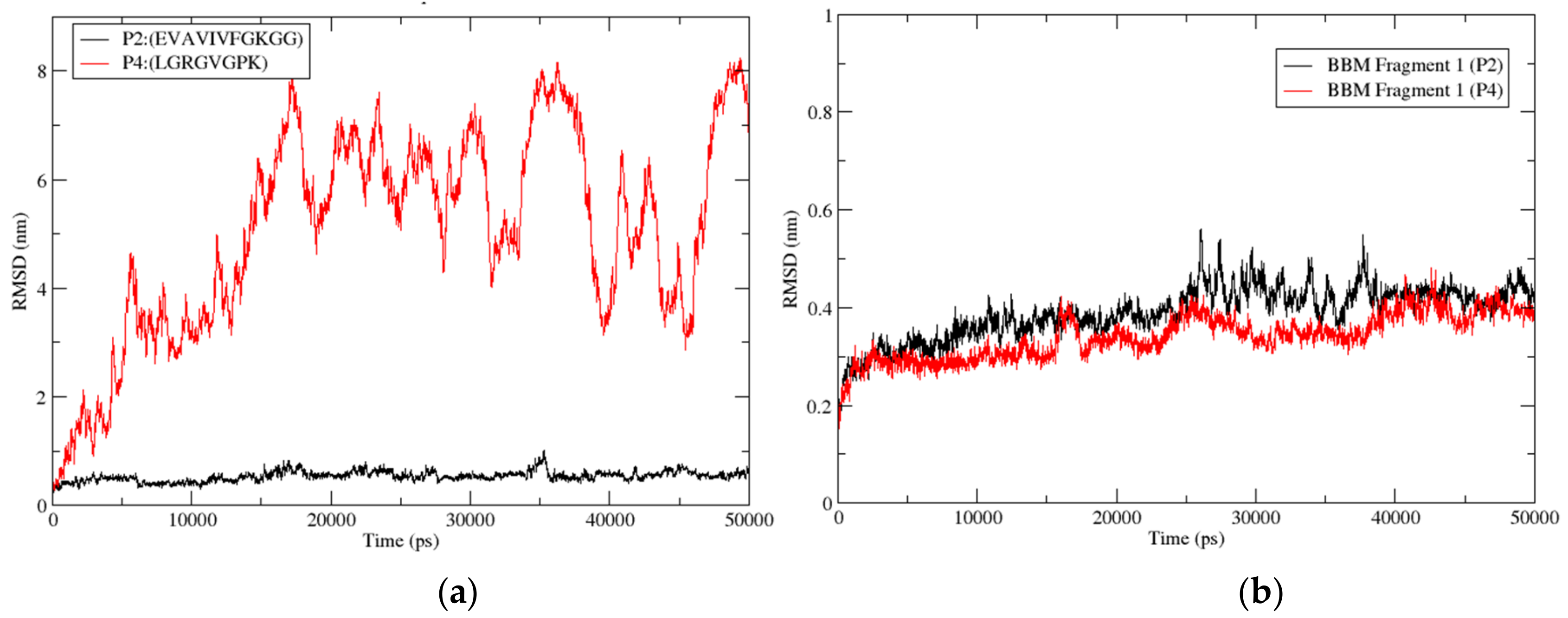
3.3.9. Root Mean Square Deviation (RMSD)
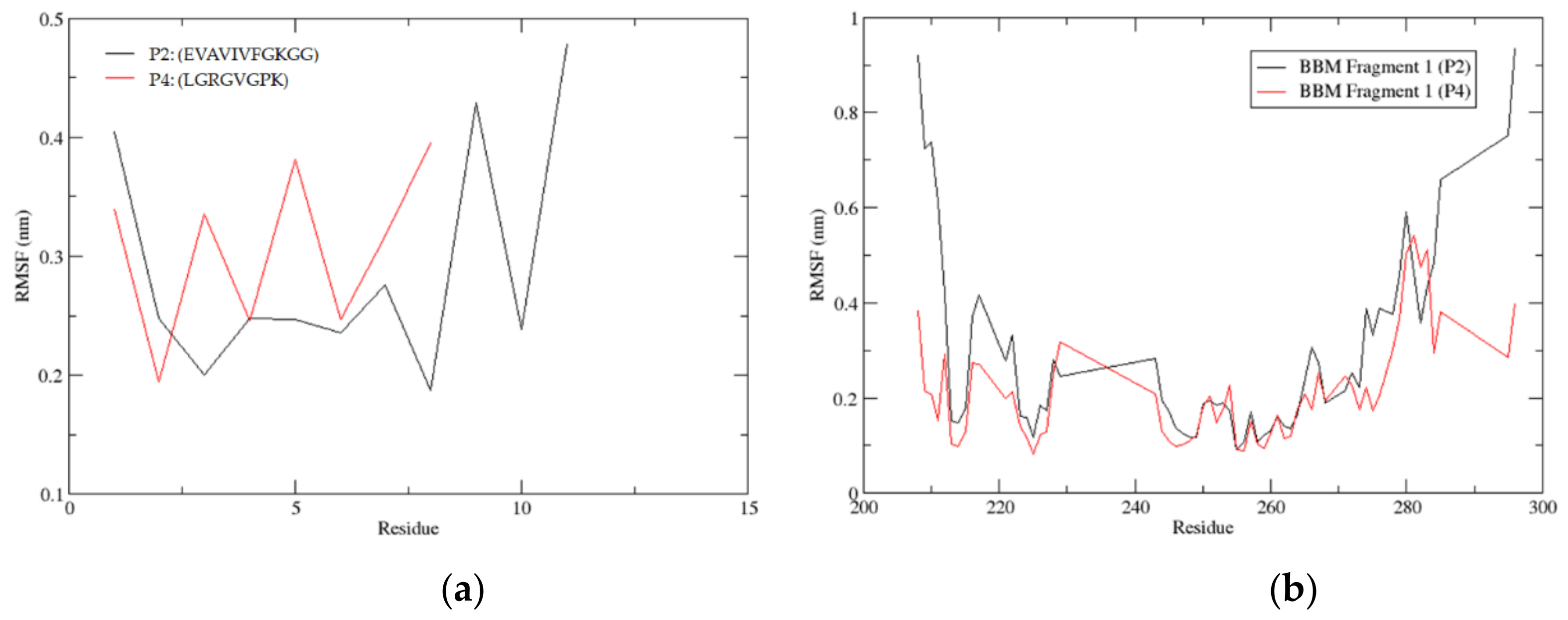
3.3.10. Root Mean Square Fluctuation (RMSF)
3.3.11. Binding Energy (Simulation Best Cluster)
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jaganathan, D.; Ramasamy, K.; Sellamuthu, G.; Jayabalan, S.; Venkataraman, G. CRISPR for crop improvement: An update review. Front. Plant Sci. 2018, 9, 985. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Zhang, D.; Shi, J. CRISPR/Cas systems: Opportunities and challenges for crop breeding. Plant Cell Rep. 2021, 40, 979–998. [Google Scholar] [CrossRef]
- Gao, C. Genome engineering for crop improvement and future. Cell 2021, 184, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Miladinovic, D.; Antunes, D.; Yildirim, K.; Bakhsh, A.; Cvejić, S.; Kondić-Špika, A.; Marjanovic Jeromela, A.; Opsahl-Sorteberg, H.G.; Zambounis, A.; Hilioti, Z. Targeted plant improvement through genome. Plant Cell Rep. 2021, 40, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, P.; Konkin, D.; Polowick, P.; Hodgins, C.L.; Subedi, M.; Xiang, D.; Yu, B.; Patterson, N.; Rajagopalan, N.; Babic, V.; et al. CRISPR/Cas9 gene editing in legume crops: Opportunities and challenges. Legume Sci. 2021, 3, e96. [Google Scholar] [CrossRef]
- Taski-Ajdukovic, K.; Nagl, N.; Miladinovic, D. Towards reducing genotype specificity in regeneration protocols after somatic hybridization between cultivated sunflowerand wild Helianthus species. Acta Biol. Hung. 2010, 61, 214–223. [Google Scholar] [CrossRef]
- Boutigny, A.L.; Dohin, N.; Pornin, D.; Rolland, M. Overview and detectability of the genetic modifications in ornamental plants. Hortic. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Bessoltane, N.; Charlot, F.; Guyon-Debast, A.; Charif, D.; Mara, K.; Collonnier, C.; Perroud, P.F.; Tepfer, M.; Nogué, F. Genome-wide specificity of plant genome editing by both CRISPR–Cas9 and TALEN. Sci. Rep. 2022, 12, 9330. [Google Scholar] [CrossRef]
- Botella, J.R. Now for the hard ones: Is there a limit on CRISPR genome editing in crops? J. Exp. Bot. 2019, 70, 734–737. [Google Scholar] [CrossRef] [Green Version]
- Lowe, K.; Wu, E.; Wang, N.; Hoerster, G.; Hastings, C.; Cho, M.J.; Scelonge, C.; Lenderts, B.; Chamberlin, M.; Cushatt, J.; et al. Morphogenic Regulators Baby boom and Wuschel Improve Monocot Transformation. Plant Cell 2016, 28, 1998–2015. [Google Scholar] [CrossRef]
- Nalapalli, S.; Tunc-Ozdemir, M.; Sun, Y.; Elumalai, S.; Que, Q. Morphogenic Regulators and Their Application in Improving Plant Transformation. In Rice Genome Engineering and Gene Editing; Bandyopadhyay, A., Thilmony, R., Eds.; Humana: New York, NY, USA, 2021; Volume 2238, pp. 37–61. [Google Scholar]
- Li, T.; Hu, J.; Sun, Y.; Li, B.; Zhang, D.; Li, W.; Liu, J.; Li, D.; Gao, C.; Zhang, Y.; et al. Highly efficient heritable genome editing in wheat using an RNA virus and bypassing tissue culture. Mol. Plant 2021, 14, 1787–1798. [Google Scholar] [CrossRef]
- Zhang, Y.; Iaffaldano, B.; Qi, Y. CRISPR ribonucleoprotein-mediated genetic engineering in plants. Plant Commun. 2021, 2, 100168. [Google Scholar] [CrossRef]
- Kumari, R.; Singh, D.P. Nano-Biotechnological Approach of Plant Genetic Engineering. In Policy Issues in Genetically Modified Crops-Global Perspectives; Singh, P., Singh, A.A., Singh, K.K., Borthakur, A., Kumar, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 481–494. [Google Scholar]
- Debernardi, J.M.; Tricoli, D.M.; Ercoli, M.F.; Hayta, S.; Ronald, P.; Palatnik, J.F.; Dubcovsky, J. A GRF-GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat. Biotechnol. 2020, 38, 1274–1279. [Google Scholar] [CrossRef]
- Lian, Z.; Nguyen, C.D.; Liu, L.; Wang, G.; Chen, J.; Wang, S.; Yi, G.; Wilson, S.; Ozias-Akins, P.; Gong, H.; et al. Application of developmental regulators to improve in planta or in vitro transformation in plants. Plant Biotechnol. J. 2022, 20, 1622–1635. [Google Scholar] [CrossRef]
- Maher, M.F.; Nasti, R.A.; Vollbrecht, M.; Starker, C.G.; Clark, M.D.; Voytas, D.F. Plant gene editing through de novo induction of meristems. Nat. Biotechnol. 2020, 38, 84–89. [Google Scholar] [CrossRef]
- Pan, W.; Cheng, Z.; Han, Z.; Yang, H.; Zhang, W.; Zhang, H. Efficient genetic transformation and CRISPR/Cas9-mediated genome editing of watermelon assisted by genes encoding developmental regulators. J. Zhejiang Univ.-Sci. B 2022, 23, 339–344. [Google Scholar] [CrossRef]
- Mujib, A.; Bansal, Y.; Malik, M.Q.; Syeed, R.; Mamgain, J.; Ejaz, B. Internal and External Regulatory Elements Controlling Somatic Embryogenesis in Catharanthus: A Model Medicinal Plant. In Somatic Embryogenesis; Ramírez-Mosqueda, M.A., Ed.; Humana: New York, NY, USA, 2022; Volume 2527, pp. 11–27. [Google Scholar]
- Deng, W.; Luo, K.; Li, Z.; Yang, Y. A Novel Method for Induction of Plant Regeneration via Somatic Embryogenesis. Plant Sci. 2009, 177, 43–48. [Google Scholar] [CrossRef]
- Laforest, L.C.; Nadakuduti, S.S. Advances in Delivery Mechanisms of CRISPR Gene-Editing Reagents in Plants. Front. Genome Ed. 2022, 4, 830178. [Google Scholar] [CrossRef]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.M.; van Lammeren, A.A.; Miki, B.L.; et al. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 2002, 14, 1737–1749. [Google Scholar] [CrossRef] [Green Version]
- Mookkan, M.; Nelson-Vasilchik, K.; Hague, J.; Zhang, Z.J.; Kausch, A.P. Selectablemarker independent transformation of recalcitrant maize inbred B73 and sorghumP898012 mediated by morphogenic regulators BABY BOOM and WUSCHEL2. Plant Cell Rep. 2017, 36, 1477–1491. [Google Scholar] [CrossRef]
- Bally, J.; Jung, H.; Mortimer, C.; Naim, F.; Philips, J.G.; Hellens, R.; Bombarely, A.; Goodin, M.M.; Waterhouse, P.M. The Rise and Rise of Nicotiana benthamiana: A Plant for All Reasons. Annu. Rev. Phytopathol. 2018, 56, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Kumar, V. BABY BOOM (BBM): A candidate transcription factor gene in plant biotechnology. Biotechnol. Lett. 2018, 40, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Maren, N.A.; Ranney, T.G.; Liu, W. New opportunities for using WUS/BBM and GRF-GIF genes to enhance genetic transformation of ornamental plants. Ornam. Plant Res. 2022, 2, 4. [Google Scholar] [CrossRef]
- Hooghvorst, I.; Nogués, S. Chromosome doubling methods in doubled haploid and haploid inducer-mediated genome-editing systems in major crops. Plant Cell Rep. 2021, 40, 255–270. [Google Scholar] [CrossRef]
- Chen, B.; Maas, L.; Figueiredo, D.; Zhong, Y.; Reis, R.; Li, M.; Horstman, A.; Riksen, T.; Weemen, M.; Liu, H.; et al. BABY BOOM regulates early embryo and endospermdevelopment. Proc. Natl. Acad. Sci. USA 2022, 119, e2201761119. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acid Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34 (Suppl. 1), D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Youkharibache, P.; Zhang, D.; Lanczycki, C.J.; Geer, R.C.; Madej, T.; Phan, L.; Ward, M.; Lu, S.; Marchler, G.H.; et al. iCn3D, a Web-based 3D Viewer for Sharing 1D/2D/3D Representations of Biomolecular Structures. Bioinformatics 2020, 36, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Vangone, A.; Bonvin, A.M. PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio-Protocol 2017, 7, e2124. [Google Scholar] [CrossRef]
- Passarinho, P.; Ketelaar, T.; Xing, M.; van Arkel, J.; Maliepaard, C.; Hendriks, M.W.; Joosen, R.; Lammers, M.; Herdies, L.; Den Boer, B.; et al. BABY BOOM target genes provide diverse entry points into cell proliferation and cell growth pathways. Plant Mol. Biol. 2008, 68, 225–237. [Google Scholar] [CrossRef]
- Ranade, S.S.; Egertsdotter, U. In silico characterization of putative gene homologues involved in somatic embryogenesis suggests that some conifer species may lack LEC2, one of the key regulators of initiation of the process. BMC Genom. 2021, 22, 392. [Google Scholar] [CrossRef]
- Wang, H.; Li, K.; Sun, X.; Xie, Y.; Han, X.; Zhang, S. Isolation and characterization of larch BABY BOOM2 and its regulation of adventitious root development. Gene 2019, 690, 90–98. [Google Scholar] [CrossRef]
- Panchangam, S.S.; Mallikarjuna, N.; Gaur, P.M.; Suravajhala, P. Androgenesis in chickpea: Anther culture and expressed sequence tags derived annotation. Indian J. Exp. Biol. 2014, 52, 181–188. [Google Scholar] [PubMed]
- Thouly, C.; Le Masson, M.; Lai, X.; Carles, C.C.; Vachon, G. Unwinding BRAHMA Functions in Plants. Genes 2020, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.; Petrey, D.; Honig, B. A hybrid method for protein–protein interface prediction. Protein Sci. 2016, 25, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Demény, M.; Ambrus, V.; Király, S.B.; Kurtán, T.; Gatti-Lafranconi, P.; Fuxreiter, M. Specific and Fuzzy Interactions Cooperate in Modulating Protein Half-Life. J. Mol. Biol. 2019, 431, 1700–1707. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Moon, Y.H.; Kang, H.G.; Jung, J.Y.; Jeon, J.S.; Sung, S.K.; An, G. Determination of the motif responsible for interaction between the rice APETALA1/AGAMOUS-LIKE9 family proteins using a yeast two-hybrid system. Plant Physiol. 1999, 120, 1193–1204. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | UniProt ID | Score |
|---|---|---|
| AGLI5 | Q38847 | 0.901 |
| LEC1 | Q9SFD8 | 0.900 |
| WUS | Q9SB92 | 0.908 |
| LEC2 | Q1PFR7 | 0.903 |
| Receptor (BBM Protein) | Ligand (Interacting Proteins) | Hex Docked Score |
|---|---|---|
| BBM-frag1 | LEC2 | −670.7 |
| BBM-frag1 | WUS | −624.5 |
| BBM-frag1 | LEC1 | −722.1 |
| BBM-frag1 | AGLI5-frag1 | −754.3 |
| BBM-frag1 | AGLI5-frag2 | −464.0 |
| Average score | −647.1 | |
| BBM-frag2 | LEC2 | −554.0 |
| BBM-frag2 | WUS | −600.9 |
| BBM-frag2 | LEC1 | −607.1 |
| BBM-frag2 | AGLI5-frag1 | −795.3 |
| BBM-frag2 | AGLI5-frag2 | −506.2 |
| Average score | −612.7 | |
| Receptor (BBM Protein) | Ligand (Interacting Proteins) | Central Structure | Binding Affinity ΔG (kcal mol−1) | Dissociation Constant Kd (M) | Polar: Polar | Non-Polar: Non-Polar |
|---|---|---|---|---|---|---|
| BBM-frag1 | LEC2 | dock46 | −11.2 | 5.7 × 10−9 | 5 | 12 |
| BBM-frag1 | WUS | dock45 | −10.6 | 1.7 × 10−8 | 0 | 9 |
| BBM-frag1 | LEC1 | dock3 | −12.3 | 8.8 × 10−10 | 4 | 11 |
| BBM-frag1 | AGLI5-frag1 | dock86 | −11.1 | 7.7 × 10−9 | 3 | 9 |
| BBM-frag1 | AGLI5-frag2 | dock28 | −10.1 | 4.0 × 10−8 | 0 | 3 |
| BBM-frag2 | LEC2 | dock28 | −8.6 | 4.8 × 10−7 | 0 | 7 |
| BBM-frag2 | WUS | dock7 | −9.5 | 1.0 × 10−7 | 0 | 15 |
| BBM-frag2 | LEC1 | dock33 | −8.7 | 4.1 × 10−7 | 2 | 32 |
| BBM-frag2 | AGLI5-frag1 | dock61 | −9.7 | 7.5 × 10−8 | 0 | 31 |
| BBM-frag2 | AGLI5-frag2 | dock2 | −8.5 | 5.9 × 10−7 | 5 | 14 |
| Residue Number | Length |
|---|---|
| LEC2 | |
| K173, E174, X, K176, N177, S178 | 6 |
| L183, X, R-185, X, V-187, X, P-189, K190 | 8 |
| W226, X, N228, N229, X, S231, X, M223 | 8 |
| I250 | 1 |
| WUS | |
| R38, W39, T40, P41, X, T43 | 6 |
| I46 | 1 |
| K50 | 1 |
| Y54 | 1 |
| W87 | 1 |
| N90, H91 | 2 |
| R94, E95 | 2 |
| LEC1 | |
| R71, I72, M73, X, K75, T76, X, P78 | 8 |
| I91 | 1 |
| V95 | 1 |
| Y98 | 1 |
| I123 | 1 |
| M127 | 1 |
| G131, F132, D133, N134, Y135, X, D137, P138, L139, X, V141, F142 | 12 |
| R145 | 1 |
| AGLI5-fragment 1 | |
| I8, K9, R10, I11 | 4 |
| R17 | 1 |
| F21 | 1 |
| R24 | 1 |
| L28 | 1 |
| L35 | 1 |
| E42, V43, A44, V45, I46, V47, F48, X, K50, X, G52 | 11 |
| T65 | 1 |
| AGLI5-fragment 2 | |
| R132 | 1 |
| K135, E136 | 2 |
| L139, T140, X, Q142, L143, E144 | 6 |
| R147 | 1 |
| E150, Q151 | 2 |
| E154, L155 | 2 |
| E158 | 1 |
| R161 | 1 |
| Residue Numbers | Length |
|---|---|
| LEC2 | |
| K173, E174, X, K176 | 4 |
| V187, X, P189, K190, R191 | 5 |
| N228, N229, X, S231, X, M233 | 6 |
| WUS | |
| R38, W39, X, P41 | 4 |
| K82, N83, X, F85, Y86, W87, X, Q89, N90 | 9 |
| R94 | 1 |
| LEC1 | |
| Y63, M64, P65 | 3 |
| N68 | 1 |
| R71, I72 | 2 |
| K75, T76 | 2 |
| V95 | 1 |
| Y98, I99 | 2 |
| T103 | 1 |
| I118 | 1 |
| A120 | 1 |
| I123 | 1 |
| M127 | 1 |
| Y135 | 1 |
| L139 | 1 |
| F142, I143 | 2 |
| Y146 | 1 |
| AGLI5-fragment 1 | |
| I8, K9, R10, I11 | 4 |
| R17 | 1 |
| F21 | 1 |
| R24 | 1 |
| L28 | 1 |
| K31 | 1 |
| L35 | 1 |
| V43, A44, V45, I46, V47, F48, X, K50 | 8 |
| AGLI5-fragment 2 | |
| L97 | 1 |
| H101 | 1 |
| L104, Q105 | 2 |
| Q123, L124, X, H126, A127 | 5 |
| T130, V131 | 2 |
| R134, K135 | 2 |
| L138, L139 | 2 |
| Q142 | 1 |
| Peptides | Docked Score | Central Cluster | Binding Affinity ΔG (kcal mol−1) | Dissociation Constant Kd (M) | Polar: Polar | Nonpolar: Nonpolar |
|---|---|---|---|---|---|---|
| P1 | −470.9 | dock16 | −7.1 | 6.0 × 10−6 | 0 | 6 |
| P2 | −379.1 | dock89 | −7.7 | 2.1 × 10−6 | 0 | 20 |
| dock10 | −7.2 | 5.4 × 10−6 | 0 | 22 | ||
| P3 | −445.5 | dock49 | −8.0 | 1.5 × 10−6 | 3 | 11 |
| P4 | −438.4 | dock96 | −8.1 | 1.1 × 10−6 | 0 | 5 |
| P5 | −366.4 | dock57 | −7.7 | 2.4 × 10−6 | 0 | 10 |
| P6 | −434.9 | dock2 | −6.7 | 1.2 × 10−5 | 0 | 15 |
| P7 | −351.1 | dock30 | −4.9 | 2.4 × 10−4 | 0 | 14 |
| Peptides | Central Cluster | Overlapping Residues between DNA-Binding Site (210–276) and Docked Interface | Length | Overlapping Residues between DNA-Binding Site (312–370) and Docked Interface | Length |
|---|---|---|---|---|---|
| P1 | dock16 | ILE210, TYR211, LEU246, GLY247, TYR249, LYS251, GLU253, LYS254, ARG257, ALA258, LEU261, ALA262, PHE275 | 13 | - | - |
| P2 | dock89 | ARG216, TYR223, ASP250, LYS251, GLU252, GLU253, LYS254, ARG257, PHE275, PRO276 | 10 | - | - |
| dock10 | ALA225, TYR245, LEU246, GLY247, GLY248, TYR249, GLU253, LYS254, ARG257, ALA258, TYR259, LEU261 | 12 | - | - | |
| P3 | dock49 | - | - | VAL316, THR317, TRP325, GLN326, ALA327, ARG328, GLN346, GLU347, ALA350, GLU351, TYR353, ASP354 | 12 |
| P4 | dock96 | ARG222, GLY248, TYR249, ASP250, LYS251, LYS254, ALA255, ARG257 | 8 | - | - |
| P5 | dock57 | TYR249, ASP250, LYS251, GLU253, LYS254, ARG257, ALA258, LEU261 | 8 | - | - |
| P6 | dock2 | TYR249, ASP250, LYS251, LYS254, ARG257, ALA258, LEU261, LEU264, LYS265, GLY268 | 10 | - | - |
| P7 | dock30 | THR317, TRP325, GLU351, ASP354, VAL316, GLN346, TYR353, ARG328, ALA349, ILE358, ALA350, ALA357 | 12 |
| Protein-Protein Complex | ΔG (kcal mol−1) | Kd (M) at 25.0 °C |
|---|---|---|
| Cluster-P2 (dock10) | −6.9 | 9.1 × 10−6 |
| Cluster-P4 | −6.1 | 3.5 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panchangam, S.S. BabyBoom: 3-Dimensional Structure-Based Ligand and Protein Interaction Prediction by Molecular Docking. Biomolecules 2022, 12, 1633. https://doi.org/10.3390/biom12111633
Panchangam SS. BabyBoom: 3-Dimensional Structure-Based Ligand and Protein Interaction Prediction by Molecular Docking. Biomolecules. 2022; 12(11):1633. https://doi.org/10.3390/biom12111633
Chicago/Turabian StylePanchangam, Sameera Sastry. 2022. "BabyBoom: 3-Dimensional Structure-Based Ligand and Protein Interaction Prediction by Molecular Docking" Biomolecules 12, no. 11: 1633. https://doi.org/10.3390/biom12111633
APA StylePanchangam, S. S. (2022). BabyBoom: 3-Dimensional Structure-Based Ligand and Protein Interaction Prediction by Molecular Docking. Biomolecules, 12(11), 1633. https://doi.org/10.3390/biom12111633