
A Pan-Draft Metabolic Model Reflects Evolutionary Diversity across 332 Yeast Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Proteomes for 332 Yeast Species
2.2. Reconstruction of Draft GEMs Using the RAVEN Toolbox
2.3. Reconstruction of a Pan-Draft Metabolic Model for 332 Yeast Species
2.4. Models’ Similarity Calculation
2.5. Trait Similarity Calculation
2.6. Evolutionary Distance Calculation across Yeast Species
2.7. Genotype Similarity Calculation
2.8. Statistical Analysis
3. Results
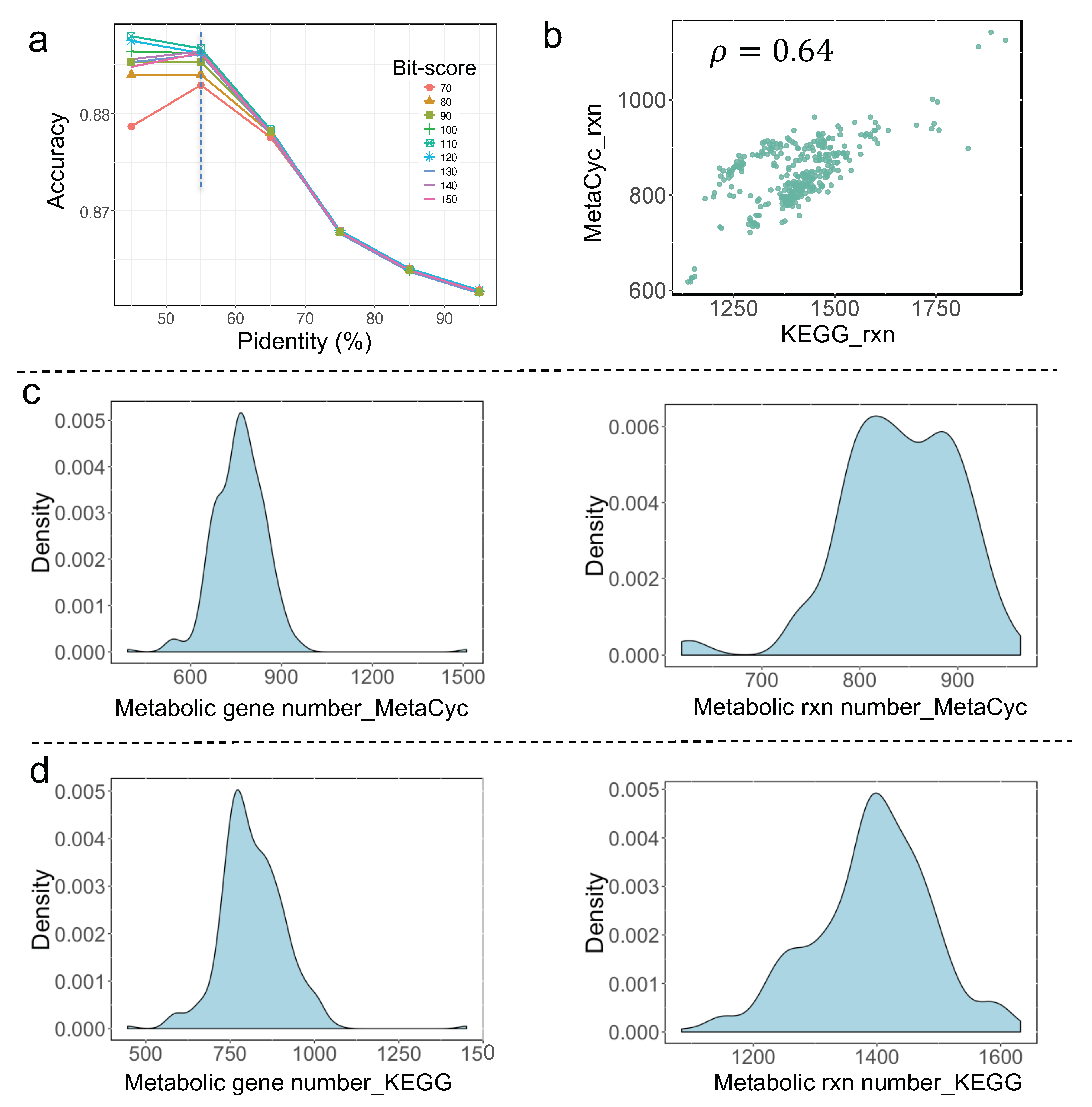
3.1. Reconstruction of Draft GEMs Using the Latest RAVEN Toolbox
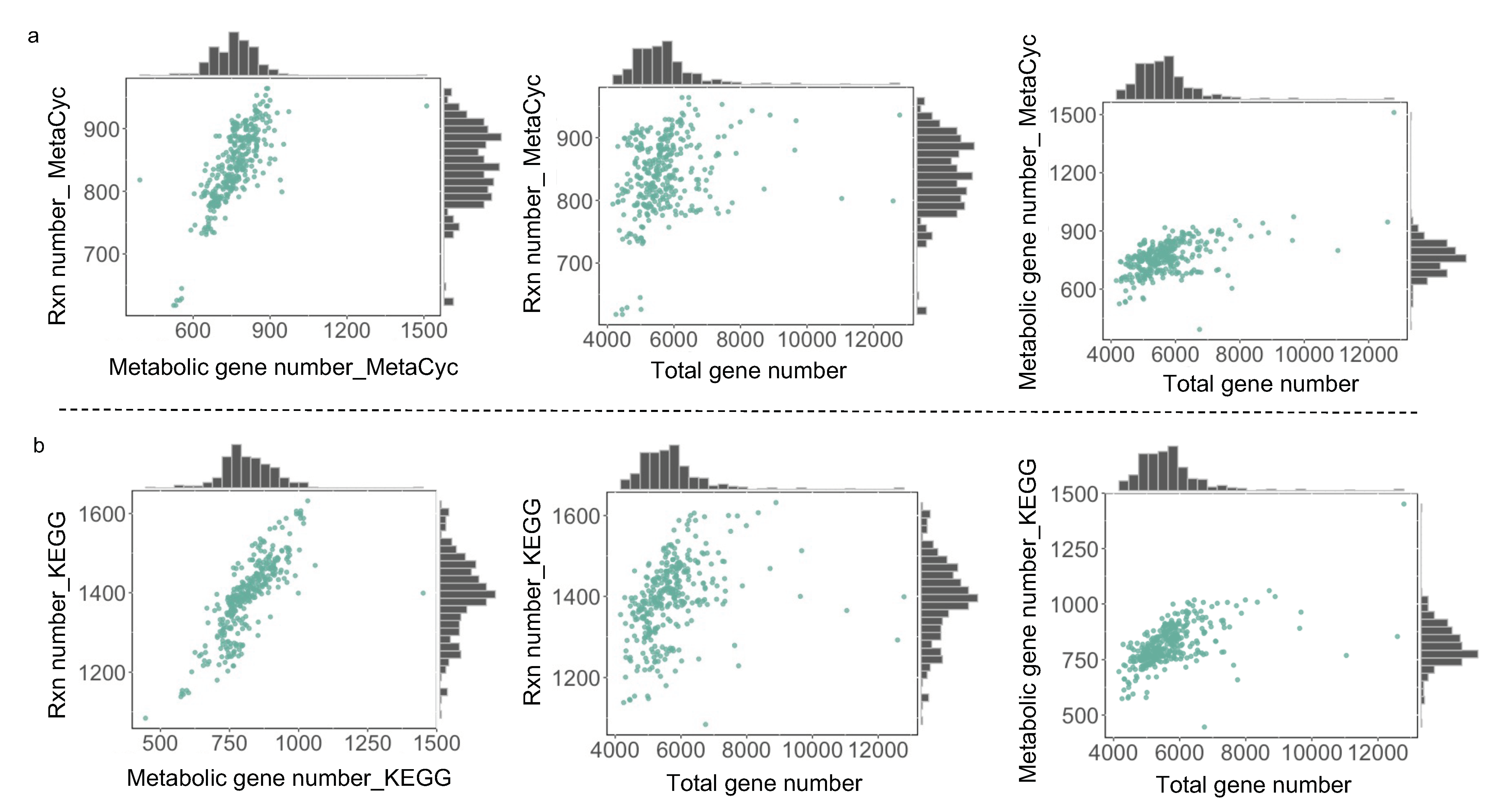
3.2. Comparative Analysis of All Draft Metabolic Models
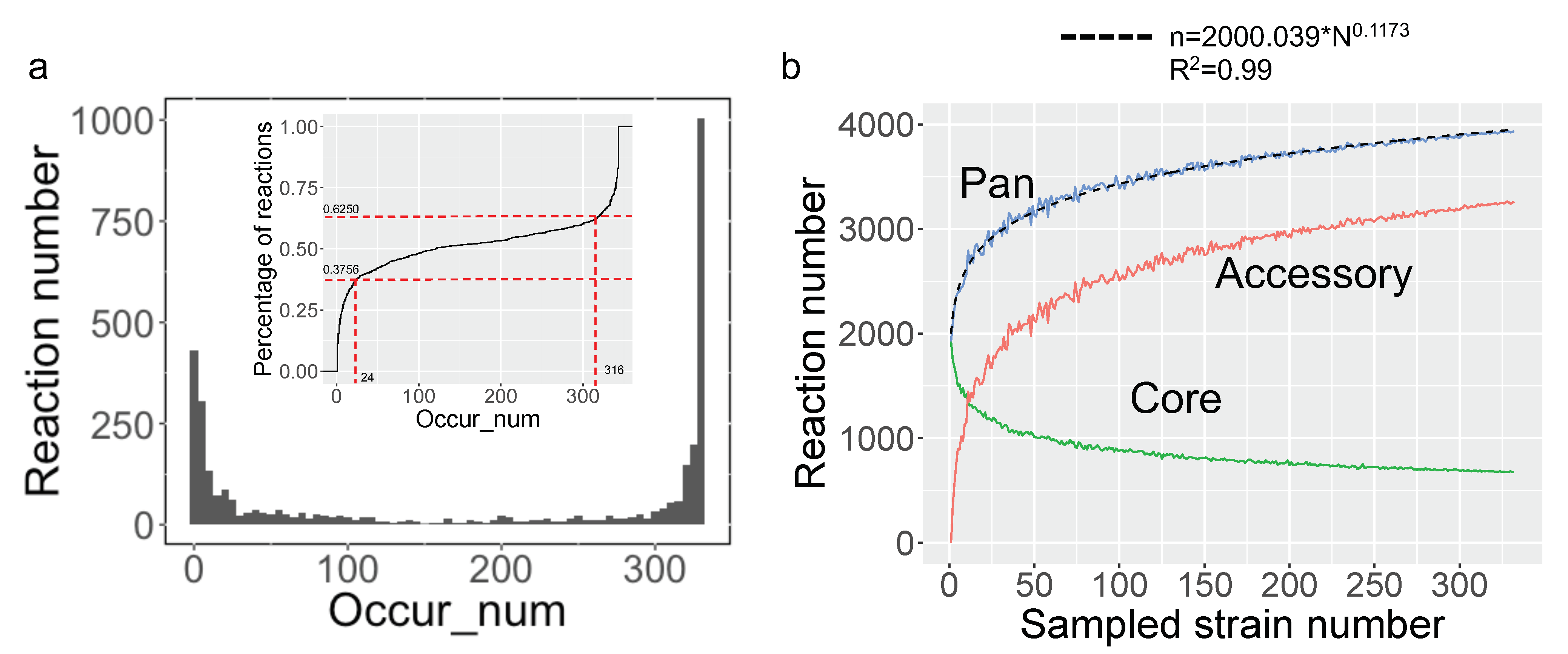
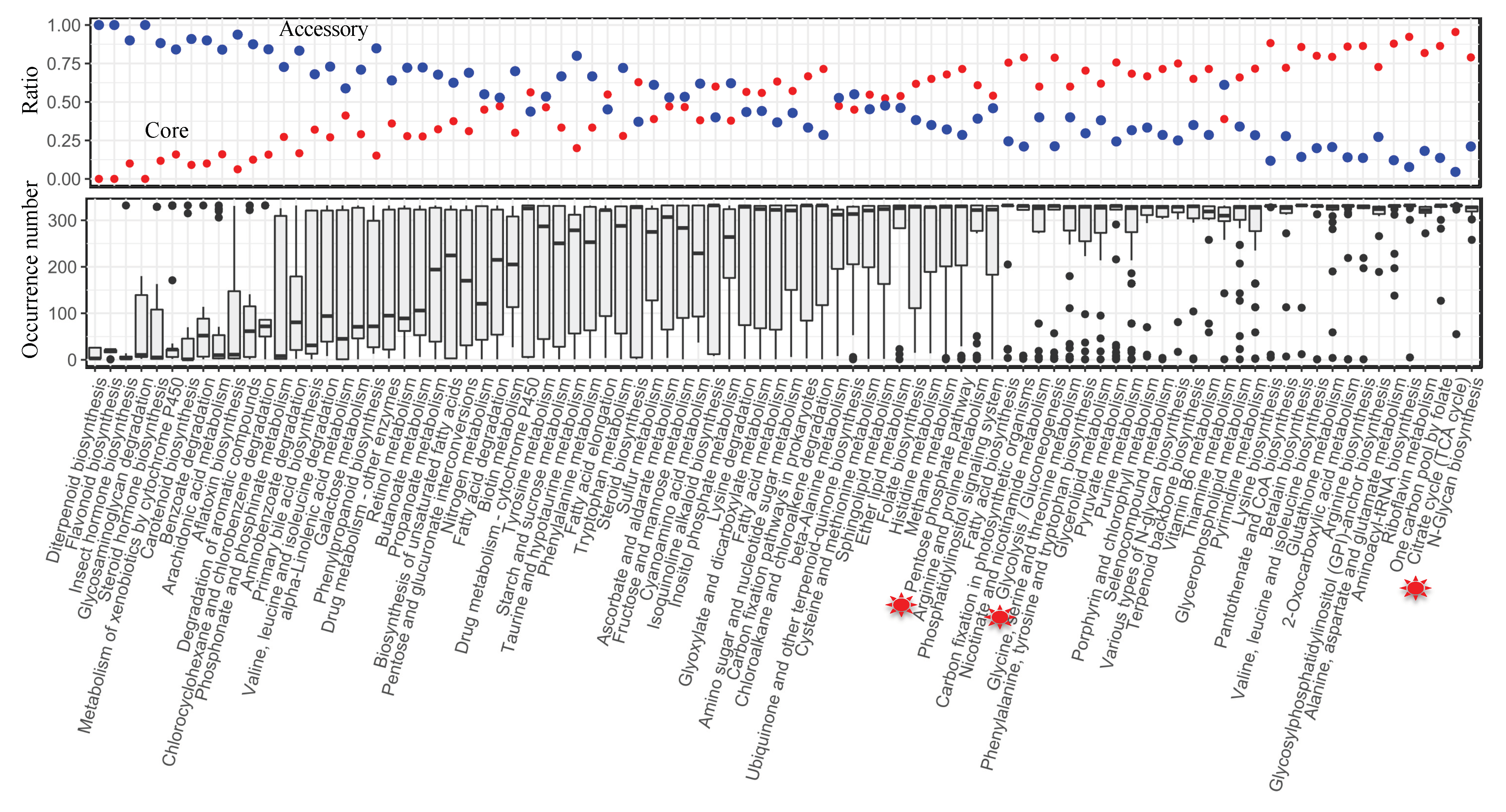
3.3. Pan-Draft Metabolic Model Reconstruction and Analysis for Budding Yeasts
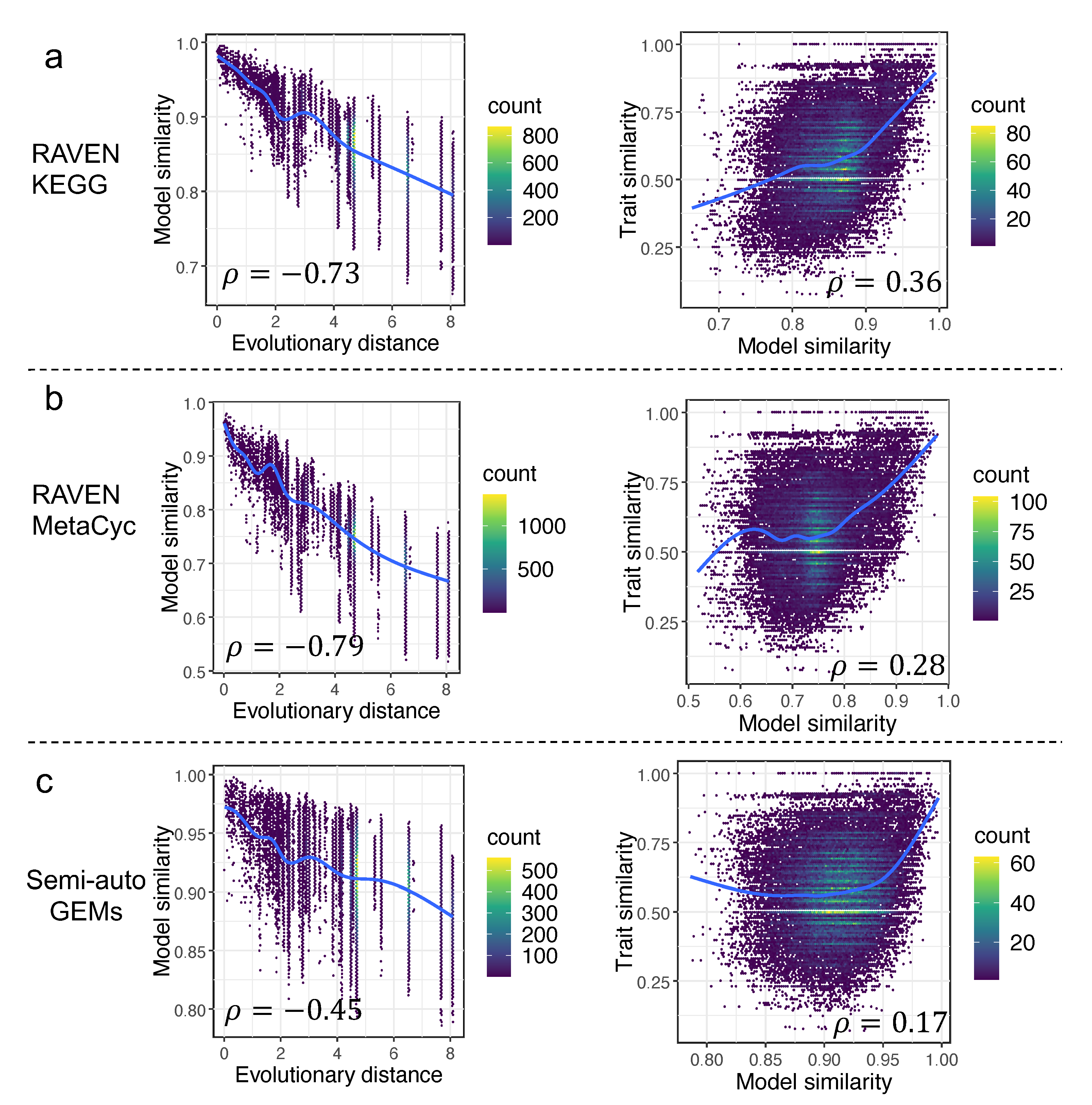
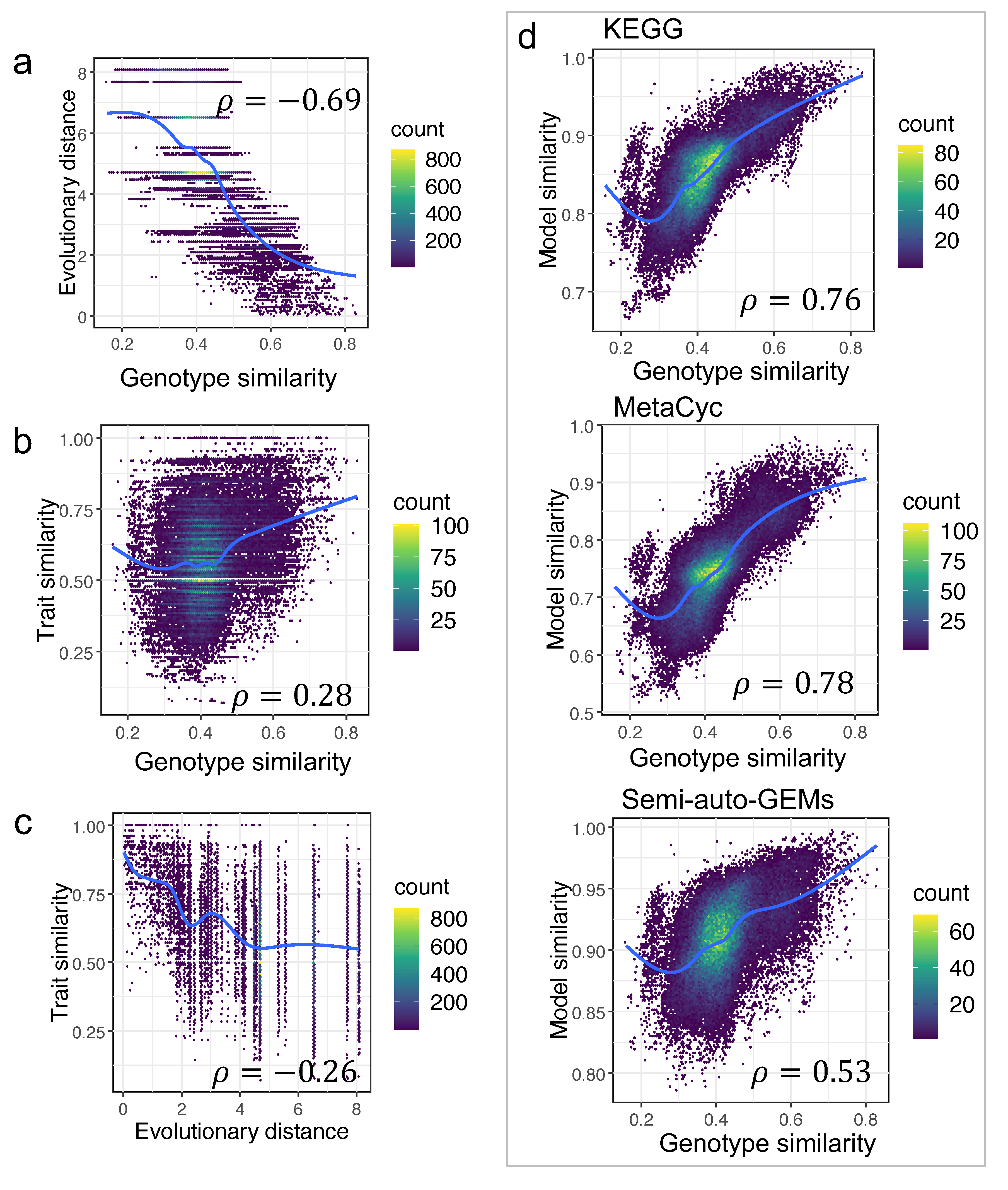
3.4. Correlations among Trait Similarity, Model Similarity, and Evolutionary Distance
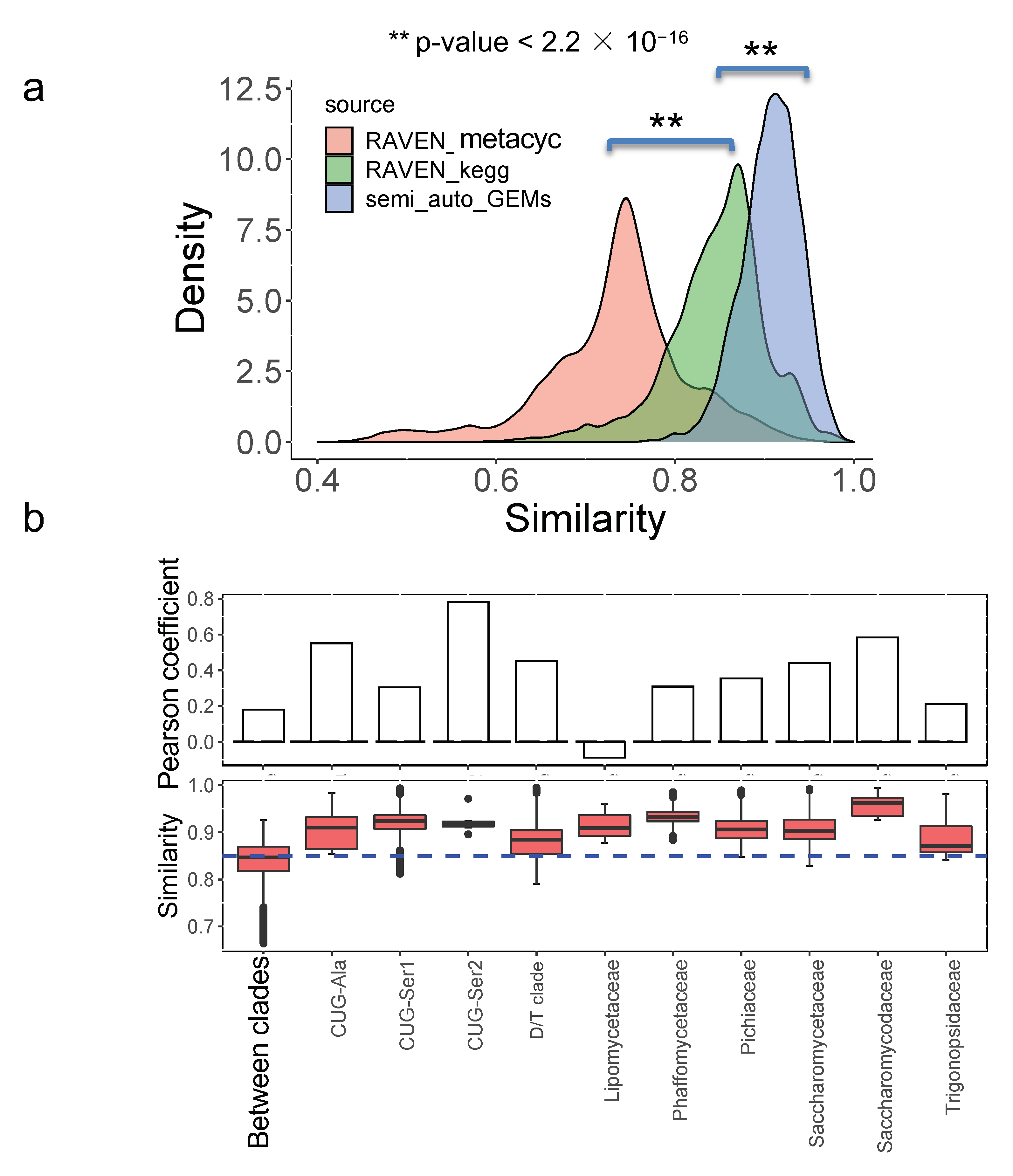
3.5. Model Similarity Comparison between Semi-Auto GEMs and Draft GEMs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, X.X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.B.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545.e20. [Google Scholar]
- Boekhout, T.; Amend, A.S.; El Baidouri, F.; Gabaldón, T.; Geml, J.; Mittelbach, M.; Robert, V.; Tan, C.S.; Turchetti, B.; Vu, D.; et al. Trends in yeast diversity discovery. Fungal. Divers. 2021, 114, 491–537. [Google Scholar]
- Dujon, B.A.; Louis, E.J. Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina). Genetics 2017, 206, 717–750. [Google Scholar]
- Patra, P.; Das, M.; Kundu, P.; Ghosh, A. Recent advances in systems and synthetic biology approaches for developing novel cell-factories in non-conventional yeasts. Biotechnol. Adv. 2021, 47, 107695. [Google Scholar]
- Mukherjee, V.; Radecka, D.; Aerts, G.; Verstrepen, K.J.; Lievens, B.; Thevelein, J.M. Phenotypic landscape of non-conventional yeast species for different stress tolerance traits desirable in bioethanol fermentation. Biotechnol. Biofuels 2017, 10, 216. [Google Scholar]
- Zhou, N.; Semumu, T.; Gamero, A. Non-Conventional Yeasts as Alternatives in Modern Baking for Improved Performance and Aroma Enhancement. Fermentation 2021, 7, 102. [Google Scholar]
- Navarrete, C.; Martínez, J.L. Non-conventional yeasts as superior production platforms for sustainable fermentation based bio-manufacturing processes. AIMS Bioeng. 2020, 7, 289–305. [Google Scholar]
- Nielsen, J. Yeast Systems Biology: Model Organism and Cell Factory. Biotechnol. J. 2019, 14, 1800421. [Google Scholar]
- Libkind, D.; Peris, D.; Cubillos, F.A.; Steenwyk, J.L.; Opulente, D.A.; Langdon, Q.K.; Rokas, A.; Hittinger, C.T. Into the wild: New yeast genomes from natural environments and new tools for their analysis. FEMS Yeast Res. 2020, 20, foaa008. [Google Scholar]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987. [Google Scholar]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010, 28, 977. [Google Scholar]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar]
- Capela, J.; Lagoa, D.; Rodrigues, R.; Cunha, E.; Cruz, F.; Barbosa, A.; Bastos, J.; Lima, D.; Ferreira, E.C.; Rocha, M.; et al. Merlin, an improved framework for the reconstruction of high-quality genome-scale metabolic models. Nucleic Acids Res. 2022, 50, 6052–6066. [Google Scholar]
- Wang, H.; Marcisauskas, S.; Sanchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0, A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158. [Google Scholar]
- Lu, H.; Kerkhoven, E.J.; Nielsen, J. Multiscale models quantifying yeast physiology: Towards a whole-cell model. Trends Biotechnol. 2021, 40, 291–305. [Google Scholar]
- Lu, H.; Li, F.; Yuan, L.; Domenzain, I.; Yu, R.; Wang, H.; Li, G.; Chen, Y.; Ji, B.; Kerkhoven, E.J.; et al. Yeast metabolic innovations emerged via expanded metabolic network and gene positive selection. Mol. Syst. Biol. 2021, 17, e10427. [Google Scholar]
- Correia, K.; Mahadevan, R. Pan-Genome-Scale Network Reconstruction: Harnessing Phylogenomics Increases the Quantity and Quality of Metabolic Models. Biotechnol. J. 2020, 15, 1900519. [Google Scholar]
- Kerkhoven, E.J.; Pomraning, K.R.; Baker, S.E.; Nielsen, J. Regulation of amino-acid metabolism controls flux to lipid accumulation in Yarrowia lipolytica. NPJ Syst. Biol. Appl. 2016, 2, 16005. [Google Scholar]
- Marcišauskas, S.; Ji, B.; Nielsen, J. Reconstruction and analysis of a Kluyveromyces marxianus genome-scale metabolic model. BMC Bioinform. 2019, 20, 551. [Google Scholar]
- Lu, H.; Li, F.; Sanchez, B.J.; Zhu, Z.; Li, G.; Domenzain, I.; Marcisauskas, S.; Anton, P.M.; Lappa, D.; Lieven, C.; et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nat. Commun. 2019, 10, 3586. [Google Scholar]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, 471–480. [Google Scholar]
- Moretti, S.; Martin, O.; Van Du Tran, T.; Bridge, A.; Morgat, A.; Pagni, M. MetaNetX/MNXref—Reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2016, 44, 523–526. [Google Scholar]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar]
- Kurtzman, C.P.; Fell, J.W.; Boekhout, T. The Yeasts: A Taxonomic Study; Elsevier Science: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Li, G.; Ji, B.; Nielsen, J. The pan-genome of Saccharomyces cerevisiae. FEMS Yeast Res. 2019, 19, foz064. [Google Scholar]
- Park, S.-C.; Lee, K.; Kim, Y.O.; Won, S.; Chun, J. Large-Scale Genomics Reveals the Genetic Characteristics of Seven Species and Importance of Phylogenetic Distance for Estimating Pan-Genome Size. Front. Microbiol. 2019, 10, 834. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Romero-Olivares, A.L.; Morrison, E.W.; Pringle, A.; Frey, S.D. Linking Genes to Traits in Fungi. Microb. Ecol. 2021, 82, 145–155. [Google Scholar]
- Plata, G.; Henry, C.S.; Vitkup, D. Long-term phenotypic evolution of bacteria. Nature 2015, 517, 369–372. [Google Scholar]
- Van der Walt, J.P. The Lipomycetaceae, a model family for phylogenetic studies. Antonie Van Leeuwenhoek 1992, 62, 247–250. [Google Scholar]
- Bernstein, D.B.; Sulheim, S.; Almaas, E.; Segre, D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol. 2021, 22, 64. [Google Scholar]
- Seif, Y.; Choudhary, K.S.; Hefner, Y.; Anand, A.; Yang, L.; Palsson, B.O. Metabolic and genetic basis for auxotrophies in Gram-negative species. Proc. Natl. Acad. Sci. USA 2020, 117, 6264–6273. [Google Scholar]
- Glasner, M.E.; Truong, D.P.; Morse, B.C. How enzyme promiscuity and horizontal gene transfer contribute to metabolic innovation. FEBS J. 2020, 287, 1323–1342. [Google Scholar]
- Ata, O.; Rebnegger, C.; Tatto, N.E.; Valli, M.; Mairinger, T.; Hann, S.; Steiger, M.G.; Calik, P.; Mattanovich, D. A single Gal4-like transcription factor activates the Crabtree effect in Komagataella phaffii. Nat. Commun. 2018, 9, 4911. [Google Scholar]
- Opulente, D.A.; Rollinson, E.J.; Bernick-Roehr, C.; Hulfachor, A.B.; Rokas, A.; Kurtzman, C.P.; Hittinger, C.T. Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol. 2018, 16, 26. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Kerkhoven, E.J.; Nielsen, J. A Pan-Draft Metabolic Model Reflects Evolutionary Diversity across 332 Yeast Species. Biomolecules 2022, 12, 1632. https://doi.org/10.3390/biom12111632
Lu H, Kerkhoven EJ, Nielsen J. A Pan-Draft Metabolic Model Reflects Evolutionary Diversity across 332 Yeast Species. Biomolecules. 2022; 12(11):1632. https://doi.org/10.3390/biom12111632
Chicago/Turabian StyleLu, Hongzhong, Eduard J. Kerkhoven, and Jens Nielsen. 2022. "A Pan-Draft Metabolic Model Reflects Evolutionary Diversity across 332 Yeast Species" Biomolecules 12, no. 11: 1632. https://doi.org/10.3390/biom12111632
APA StyleLu, H., Kerkhoven, E. J., & Nielsen, J. (2022). A Pan-Draft Metabolic Model Reflects Evolutionary Diversity across 332 Yeast Species. Biomolecules, 12(11), 1632. https://doi.org/10.3390/biom12111632