
Targeting the Endocannabinoidome in Pancreatic Cancer



Abstract
:1. Introduction
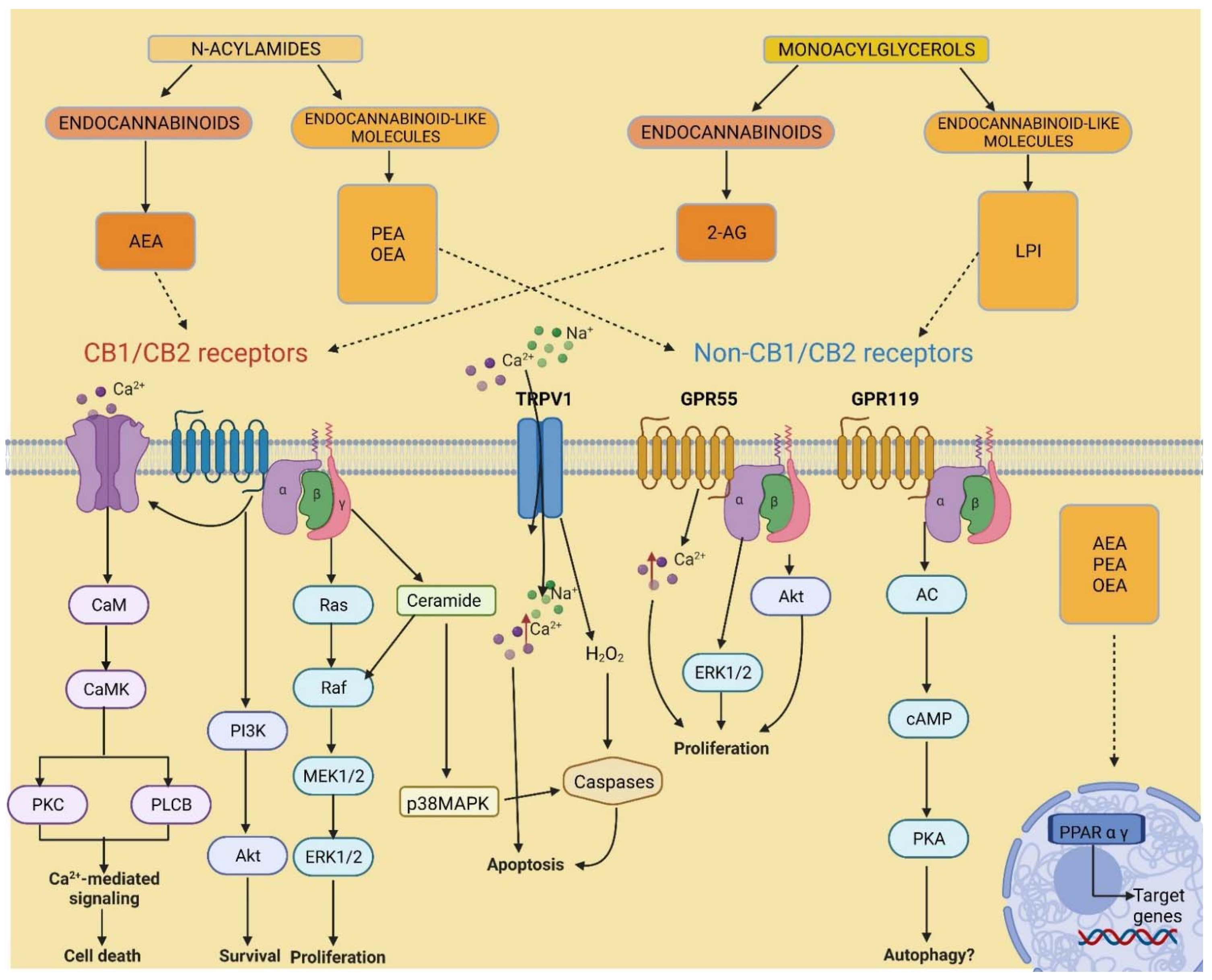
2. Endocannabinoidome
3. Pancreatic Cancer
4. Expression of Endocannabinoidome Members in Pancreatic Cancer
5. Involvement of the Endocannabinoidome in Pancreatic Cancer
5.1. Cannabinoid Receptors
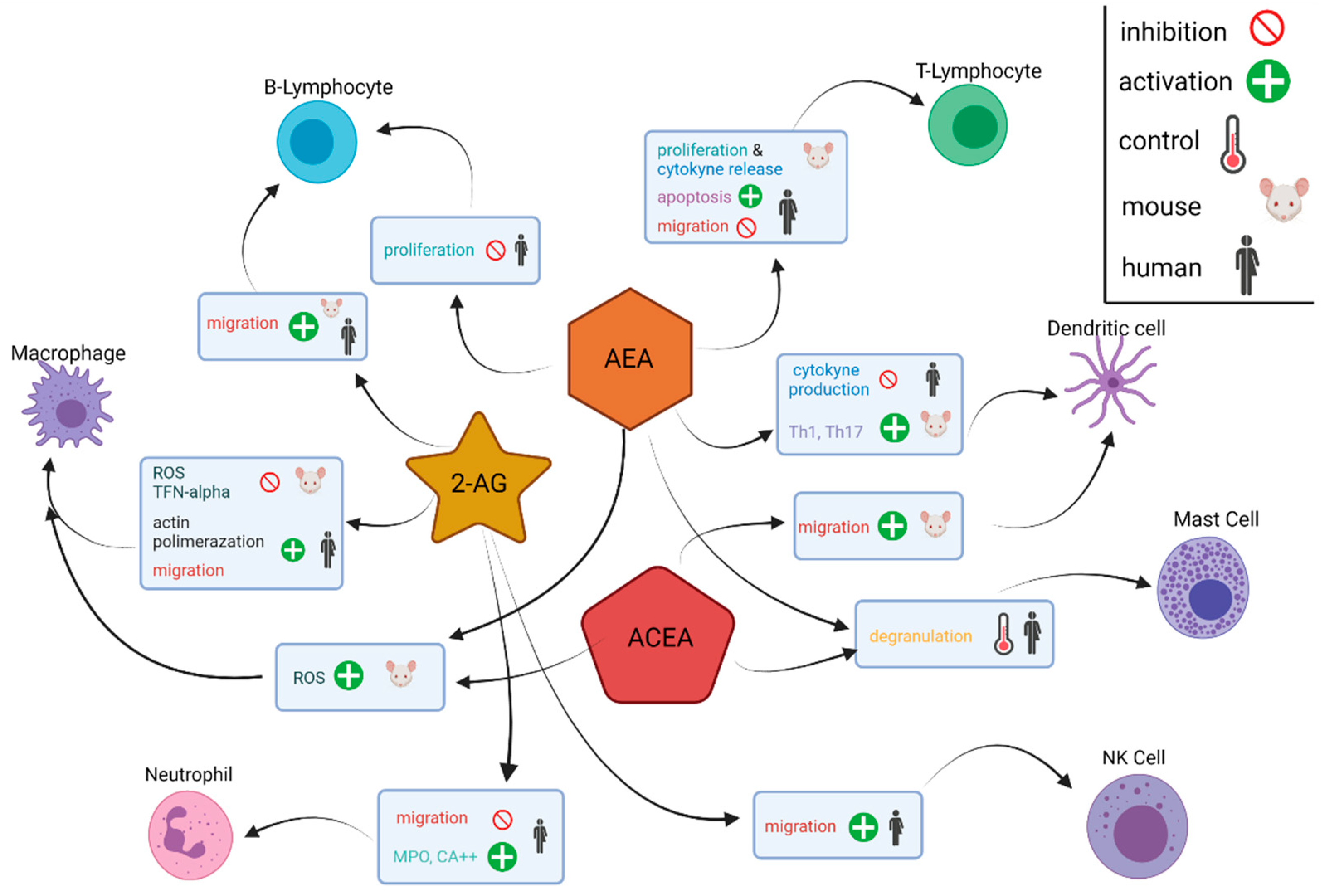
5.2. The TME and the Immune Endocannabinoid System
5.3. GPR55 Signalling
5.4. Role of Endocannabinoid Receptor Heterodimers
6. Therapeutic Opportunities
6.1. Palliative Care
6.2. Combined Chemotherapy
6.2.1. In Vitro
6.2.2. In Vivo
6.3. Cannabinoids in Combined Radiotherapy
6.4. Cannabinoids as Immunotherapy Agents
6.5. Inhibitors of Endocannabinoid-Metabolising Enzymes
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, M.; Tang, Z.; Wu, X.; Spengler, R.; Jiang, H.; Yang, Y.; Boivin, N. The origins of cannabis smoking: Chemical residue evidence from the first millennium BCE in the Pamirs. Sci. Adv. 2019, 5, eaaw1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.-E.; Li, X.; Zhao, Y.-X.; Ferguson, D.K.; Hueber, F.; Bera, S.; Wang, Y.-F.; Zhao, L.-C.; Liu, C.-J.; Li, C.-S. A new insight into Cannabis sativa (Cannabaceae) utilization from 2500-year-old Yanghai Tombs, Xinjiang, China. J. Ethnopharmacol. 2006, 108, 414–422. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Management of Substance Abuse: Cannabis. 2016. Available online: http://www.who.int/substance_abuse/facts/cannabis/en/ (accessed on 1 February 2022).
- Srebnik, M.; Lander, N.; Breuer, A.; Mechoulam, R. Base-catalysed double-bond isomerizations of cannabinoids: Structural and stereochemical aspects. J. Chem. Soc. Perkin Trans. 1984, 1, 2881–2886. [Google Scholar] [CrossRef]
- Mechoulam, R.; Hanuš, L.R. A historical overview of chemical research on cannabinoids. Chem. Phys. Lipids 2000, 108, 1–13. [Google Scholar] [CrossRef]
- Peng, H.; Shahidi, F. Cannabis and Cannabis Edibles: A Review. J. Agric. Food Chem. 2021, 69, 1751–1774. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Melck, D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Alves, V.L.; Gonçalves, J.L.; Aguiar, J.; Teixeira, H.M.; Câmara, J.S. The synthetic cannabinoids phenomenon: From structure to toxicological properties. A review. Crit. Rev. Toxicol. 2020, 50, 359–382. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharm. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Pharmacological, physiological and clinical implications of the discovery of cannabinoid receptors. Biochem. Soc. Trans. 1998, 26, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Randall, M.D.; Kendall, D.A. Endocannabinoids: A new class of vasoactive substances. Trends Pharm. Sci. 1998, 19, 55–58. [Google Scholar] [CrossRef]
- Di Marzo, V.; Deutsch, D.G. Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol. Dis. 1998, 5, 386–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maione, S.; Costa, B.; Di Marzo, V. Endocannabinoids: A unique opportunity to develop multitarget analgesics. Pain 2013, 154 (Suppl. S1), S87–S93. [Google Scholar] [CrossRef] [PubMed]
- Sharafi, G.; He, H.; Nikfarjam, M. Potential Use of Cannabinoids for the Treatment of Pancreatic Cancer. J. Pancreat Cancer 2019, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.; Farrah, K. CADTH Rapid Response Reports. In Medical Cannabis Use in Palliative Care: Review of Clinical Effectiveness and Guidelines—An Update; Canadian Agency for Drugs and Technologies in Health Copyright © 2022 Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2019. [Google Scholar]
- Malhotra, P.; Casari, I.; Falasca, M. Therapeutic potential of cannabinoids in combination cancer therapy. Adv. Biol. Regul. 2021, 79, 100774. [Google Scholar] [CrossRef]
- Mechoulam, R.; Fride, E.; Di Marzo, V. Endocannabinoids. Eur. J. Pharm. 1998, 359, 1–18. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Bari, M.; Battista, N.; Fezza, F.; Gasperi, V.; Maccarrone, M. New insights into endocannabinoid degradation and its therapeutic potential. Mini Rev. Med. Chem. 2006, 6, 257–268. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular Characterization of a Phospholipase D Generating Anandamide and Its Congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: Pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggelaar, M.P.; Maccarrone, M.; van der Stelt, M. 2-Arachidonoylglycerol: A signaling lipid with manifold actions in the brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Di Marzo, V. Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G-protein-coupled receptors and transient receptor potential channels. J. Neuroimmune Pharm. 2010, 5, 103–121. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Munawar, N.; Oriowo, M.A.; Masocha, W. Antihyperalgesic Activities of Endocannabinoids in a Mouse Model of Antiretroviral-Induced Neuropathic Pain. Front. Pharm. 2017, 8, 136. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Saito, O.; Tanabe, M.; Inayoshi, K.; Kobata, K.; Uno, S.; Morita, A.; Watanabe, T. Monoacylglycerols activate capsaicin receptor, TRPV1. Lipids 2008, 43, 471–483. [Google Scholar] [CrossRef]
- Bouaboula, M.; Hilairet, S.; Marchand, J.; Fajas, L.; Fur, G.L.; Casellas, P. Anandamide induced PPARγ transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005, 517, 174–181. [Google Scholar] [CrossRef]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol. Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef]
- Tudurí, E.; Imbernon, M.; Hernández-Bautista, R.J.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202. [Google Scholar] [CrossRef] [PubMed]
- Ghislain, J.; Poitout, V. Targeting lipid GPCRs to treat type 2 diabetes mellitus—Progress and challenges. Nat. Rev. Endocrinol. 2021, 17, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Lago-Fernandez, A.; Hurst, D.P.; Sotudeh, N.; Brailoiu, E.; Reggio, P.H.; Abood, M.E.; Jagerovic, N. Therapeutic Exploitation of GPR18: Beyond the Cannabinoids? J. Med. Chem. 2020, 63, 14216–14227. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Cordero-Morales, J.F.; Liu, B.; Qin, F.; Julius, D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 2013, 77, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Jung, D.Y.; Kim, J.H.; Patel, P.R.; Hu, X.; Lee, Y.; Azuma, Y.; Wang, H.F.; Tsitsilianos, N.; Shafiq, U.; et al. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. Faseb J. 2015, 29, 3182–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef]
- Piñeiro, R.; Falasca, M. Lysophosphatidylinositol signalling: New wine from an old bottle. Biochim. et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N.; Tsuboi, K.; Uyama, T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [Google Scholar] [CrossRef]
- Bhandari, S.; Bisht, K.S.; Merkler, D.J. The Biosynthesis and Metabolism of the N-Acylated Aromatic Amino Acids: N-Acylphenylalanine, N-Acyltyrosine, N-Acyltryptophan, and N-Acylhistidine. Front. Mol. Biosci. 2021, 8, 801749. [Google Scholar] [CrossRef]
- Di Marzo, V.; Wang, J. The Endocannabinoidome: The World of Endocannabinoids and Related Mediators; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Rahman, I.A.S.; Tsuboi, K.; Uyama, T.; Ueda, N. New players in the fatty acyl ethanolamide metabolism. Pharmacol. Res. 2014, 86, 1–10. [Google Scholar] [CrossRef]
- Iannotti, F.; Piscitelli, F.; Martella, A.; Mazzarella, E.; Allarà, M.; Palmieri, V.; Parrella, C.; Capasso, R.; Di Marzo, V. Analysis of the “endocannabinoidome” in peripheral tissues of obese Zucker rats. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 127–135. [Google Scholar] [CrossRef]
- Lian, J.; Casari, I.; Falasca, M. Modulatory role of the endocannabinoidome in the pathophysiology of the gastrointestinal tract. Pharmacol. Res. 2021, 175, 106025. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Fidler-Benaoudia, M.; Keegan, T.H.; Hipp, H.S.; Jemal, A.; Siegel, R.L. Cancer statistics for adolescents and young adults, 2020. CA A Cancer J. Clin. 2020, 70, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Sener, S.F.; Fremgen, A.; Menck, H.R.; Winchester, D.P. Pancreatic cancer: A report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J. Am. Coll. Surg. 1999, 189, 1–7. [Google Scholar] [CrossRef]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2008, 122, 742–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehn, B.M. Looking to Long-term Survivors for Improved Pancreatic Cancer Treatment. JAMA 2020, 324, 2242–2244. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: A conspiracy theory. World J. Gastroenterol. 2014, 20, 11216–11229. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.R.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Christine Cripps, M.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Adjuvant and neoadjuvant treatment in pancreatic cancer. World J. Gastroenterol. 2012, 18, 1565–1572. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research; The National Academies Press: Washington, DC, USA, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.; Arifin, S.; Fyffe, C.; Sala, G.; Sacchetto, L.; Chiorino, G. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol. Sci. 2018, 39, 586–604. [Google Scholar] [CrossRef]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Canela, E.I. The Interplay between Cancer Biology and the Endocannabinoid System-Significance for Cancer Risk, Prognosis and Response to Treatment. Cancers 2020, 12, 3275. [Google Scholar] [CrossRef]
- Hartel, M.; di Mola, F.F.; Selvaggi, F.; Mascetta, G.; Wente, M.N.; Felix, K.; Giese, N.A.; Hinz, U.; Di Sebastiano, P.; Büchler, M.W.; et al. Vanilloids in pancreatic cancer: Potential for chemotherapy and pain management. Gut 2006, 55, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, J.; Qiu, L. Transient receptor potential vanilloid 1 promotes EGFR ubiquitination and modulates EGFR/MAPK signalling in pancreatic cancer cells. Cell Biochem. Funct. 2020, 38, 401–408. [Google Scholar] [CrossRef]
- Liu, X.; Qian, D.; Liu, H.; Abbruzzese, J.L.; Luo, S.; Walsh, K.M.; Wei, Q. Genetic variants of the peroxisome proliferator-activated receptor (PPAR) signaling pathway genes and risk of pancreatic cancer. Mol. Carcinog. 2020, 59, 930–939. [Google Scholar] [CrossRef]
- Sailler, S.; Schmitz, K.; Jäger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272. [Google Scholar] [CrossRef] [Green Version]
- Nithipatikom, K.; Isbell, M.A.; Endsley, M.P.; Woodliff, J.E.; Campbell, W.B. Anti-proliferative effect of a putative endocannabinoid, 2-arachidonylglyceryl ether in prostate carcinoma cells. Prostaglandins Other Lipid Mediat. 2011, 94, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Tomar, S.; Zumbrun, E.E.; Nagarkatti, M.; Nagarkatti, P.S. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J. Pharmacol. Exp. Ther. 2015, 353, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Glogauer, J.; Blay, J. Cannabinoids, their cellular receptors, and effects on the invasive phenotype of carcinoma and metastasis. Cancer Rep. 2021, e1475. [Google Scholar] [CrossRef]
- Guzman, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef]
- Casanova, M.L.; Blázquez, C.; Martínez-Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; del Pulgar, T.G.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [Green Version]
- Brandi, J.; Dando, I.; Palmieri, M.; Donadelli, M.; Cecconi, D. Comparative proteomic and phosphoproteomic profiling of pancreatic adenocarcinoma cells treated with CB1 or CB2 agonists. Electrophoresis 2013, 34, 1359–1368. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The endocannabinoid system: A target for cancer treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [Green Version]
- Michalski, C.W.; Maier, M.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Giese, N.A.; Giese, T.; Friess, H.; et al. Cannabinoids reduce markers of inflammation and fibrosis in pancreatic stellate cells. PLoS ONE 2008, 3, e1701. [Google Scholar] [CrossRef] [PubMed]
- von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Jöhnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and local immunosuppression in pancreatic cancer patients. Clin. Cancer Res. 2001, 7, 925s–932s. [Google Scholar] [PubMed]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef]
- Iozzo, M.; Sgrignani, G.; Comito, G.; Chiarugi, P.; Giannoni, E. Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches. Cells 2021, 10, 3396. [Google Scholar] [CrossRef]
- Kienzl, M.; Kargl, J.; Schicho, R. The Immune Endocannabinoid System of the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 8929. [Google Scholar] [CrossRef]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1 and CB2 receptors in immune cells. Acta Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef]
- Qiu, C.; Yang, L.; Wang, B.; Cui, L.; Li, C.; Zhuo, Y.; Zhang, L.; Zhang, S.; Zhang, Q.; Wang, X. The role of 2-arachidonoylglycerol in the regulation of the tumor-immune microenvironment in murine models of pancreatic cancer. Biomed. Pharm. 2019, 115, 108952. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Bärnthaler, T.; Heitzer, E.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; et al. G protein-coupled receptor GPR55 promotes colorectal cancer and has opposing effects to cannabinoid receptor 1. Int. J. Cancer 2018, 142, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Smith, N.J. Allosteric modulation of heterodimeric G-protein-coupled receptors. Trends Pharm. Sci. 2007, 28, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Zoratti, C.; Osibow, K.; Balenga, N.; Goessnitzer, E.; Waldhoer, M.; Malli, R.; Graier, W.F. Integrin clustering enables anandamide-induced Ca2+ signaling in endothelial cells via GPR55 by protection against CB1-receptor-triggered repression. J. Cell Sci. 2008, 121, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balenga, N.A.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Capone, E.; Damiani, V.; Akkaya, B.G.; Linton, K.J.; Di Sebastiano, P.; Chen, X.; Keeton, A.B.; Ramirez-Alcantara, V.; et al. Pharmacological inhibition of ABCC3 slows tumour progression in animal models of pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 312. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [Green Version]
- Blasco-Benito, S.; Moreno, E.; Seijo-Vila, M.; Tundidor, I.; Andradas, C.; Caffarel, M.M.; Caro-Villalobos, M.; Urigüen, L.; Diez-Alarcia, R.; Moreno-Bueno, G.; et al. Therapeutic targeting of HER2–CB2R heteromers in HER2-positive breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3863–3872. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Gómez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; García-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martín-Villar, E.; Flores, J.M.; et al. Role of Cannabinoid Receptor CB2 in HER2 Pro-oncogenic Signaling in Breast Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef] [Green Version]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br. J. Pharm. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [Green Version]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saur, D.; Seidler, B.; Schneider, G.; Algül, H.; Beck, R.; Senekowitsch–Schmidtke, R.; Schwaiger, M.; Schmid, R.M. CXCR4 Expression Increases Liver and Lung Metastasis in a Mouse Model of Pancreatic Cancer. Gastroenterology 2005, 129, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Domenichini, A.; Edmands, J.S.; Adamska, A.; Begicevic, R.-R.; Paternoster, S.; Falasca, M. Pancreatic cancer tumorspheres are cancer stem-like cells with increased chemoresistance and reduced metabolic potential. Adv. Biol. Regul. 2019, 72, 63–77. [Google Scholar] [CrossRef]
- Lohse, I.; Wildermuth, E.; Brothers, S.P. Naturally occurring compounds as pancreatic cancer therapeutics. Oncotarget 2018, 9, 35448–35457. [Google Scholar] [CrossRef] [Green Version]
- Su, T.-F.; Zhang, L.-H.; Peng, M.; Wu, C.-H.; Pan, W.; Tian, B.; Shi, J.; Pan, H.-L.; Li, M. Cannabinoid CB2 Receptors Contribute to Upregulation of β-endorphin in Inflamed Skin Tissues by Electroacupuncture. Mol. Pain 2011, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auh, Q.-S.; Chun, Y.H.; Melemedjian, O.K.; Zhang, Y.; Ro, J.Y. Peripheral interactions between cannabinoid and opioid receptor agonists in a model of inflammatory mechanical hyperalgesia. Brain Res. Bull. 2016, 125, 211–217. [Google Scholar] [CrossRef]
- Singh, N.S.; Bernier, M.; Wainer, I.W. Selective GPR55 antagonism reduces chemoresistance in cancer cells. Pharm. Res. 2016, 111, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Nahler, G.; Likar, R. Cannabidiol Possibly Improves Survival of Patients with Pancreatic Cancer: A Case Series. Clin. Oncol. Res. 2020, 3, 1–4. [Google Scholar] [CrossRef]
- Yasmin-Karim, S.; Moreau, M.; Mueller, R.; Sinha, N.; Dabney, R.; Herman, A.; Ngwa, W. Enhancing the Therapeutic Efficacy of Cancer Treatment With Cannabinoids. Front. Oncol. 2018, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Huynh, N.; Dumesny, C.; Wang, K.; He, H.; Nikfarjam, M. Cannabinoids Inhibited Pancreatic Cancer via P-21 Activated Kinase 1 Mediated Pathway. Int. J. Mol. Sci. 2020, 21, 8035. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Wittig, F.; Hinz, B. The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies. Cancers 2021, 13, 5701. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Title | Reference | Location | Status |
|---|---|---|---|---|
| Pancreatic Neoplasm, Cachexia | The effect of Cannabis in Pancreatic Cancer | NCT03245658 | Naestved, Denmark | Unknown |
| Pancreatic Cancer Non-resectable and Metastatic, Chemotherapy-induced Nausea and Vomiting | Efficacy and safety of Dronabinol in the improvement of Chemotherapy-induced and Tumour-related Symptoms in Advanced Pancreatic Cancer | NCT03984214 | Oberösterreich, Klagenfurt, Leoben, Salzburg, St. Veit an der Glan, Steyr, Vienna, Zams Austria | Recruiting |
| Pancreatic Cancer | Nutrition and Pharmacological Algorithm for Oncology patients’ study | NCT04155008 | New York, United States | Recruiting |
| Hepatocellular Carcinoma, Pancreatic Cancer | A Study of Dexanabinol in Combination with Chemotherapy in Patients with Advanced Tumours | NCT02423239 | Germany, Poland, Spain, United Kingdom | Unknown |
| Cancer of Pancreas, Liver, Rectum, Colon, Gall Bladder | A Study of the Efficacy of Cannabidiol Patients with Multiple Myeloma, Glioblastoma Multiforme, and GI Malignancies | NCT03607643 | Florida, United States | Unknown |
| Hepatocellular Carcinoma, Cholangiocarcinoma, GastroEsophageal, Colorectal and Pancreatic cancer | TPST-1120 as Monotherapy and in Combination with Nivolumab in Subjects with Advanced Cancers | NCT03829436 | California, Florida, Maryland, Michigan, New York, North Carolina, Oklahoma, Pennsylvania, Tennessee, United States | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falasca, V.; Falasca, M. Targeting the Endocannabinoidome in Pancreatic Cancer. Biomolecules 2022, 12, 320. https://doi.org/10.3390/biom12020320
Falasca V, Falasca M. Targeting the Endocannabinoidome in Pancreatic Cancer. Biomolecules. 2022; 12(2):320. https://doi.org/10.3390/biom12020320
Chicago/Turabian StyleFalasca, Valerio, and Marco Falasca. 2022. "Targeting the Endocannabinoidome in Pancreatic Cancer" Biomolecules 12, no. 2: 320. https://doi.org/10.3390/biom12020320
APA StyleFalasca, V., & Falasca, M. (2022). Targeting the Endocannabinoidome in Pancreatic Cancer. Biomolecules, 12(2), 320. https://doi.org/10.3390/biom12020320