
Mechanistic Investigation of GHS-R Mediated Glucose-Stimulated Insulin Secretion in Pancreatic Islets

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Glucose Stimulated Insulin Secretion (GSIS) In Vivo
2.3. Immunohistochemistry of Pancreas
2.4. Islet Isolation
2.5. Glucose-Stimulated Insulin Secretion (GSIS)
2.6. Insulin Content of Pancreas and Islets
2.7. Respirometry of Islets
2.8. ATP/ADP Ratio
2.9. Glucose-6-Phosphate Measurement
2.10. Real-Time RT-PCR
2.11. Western Blot
2.12. Extraction and Quantification of Mitochondrial DNA
2.13. Statistical Analysis
3. Results
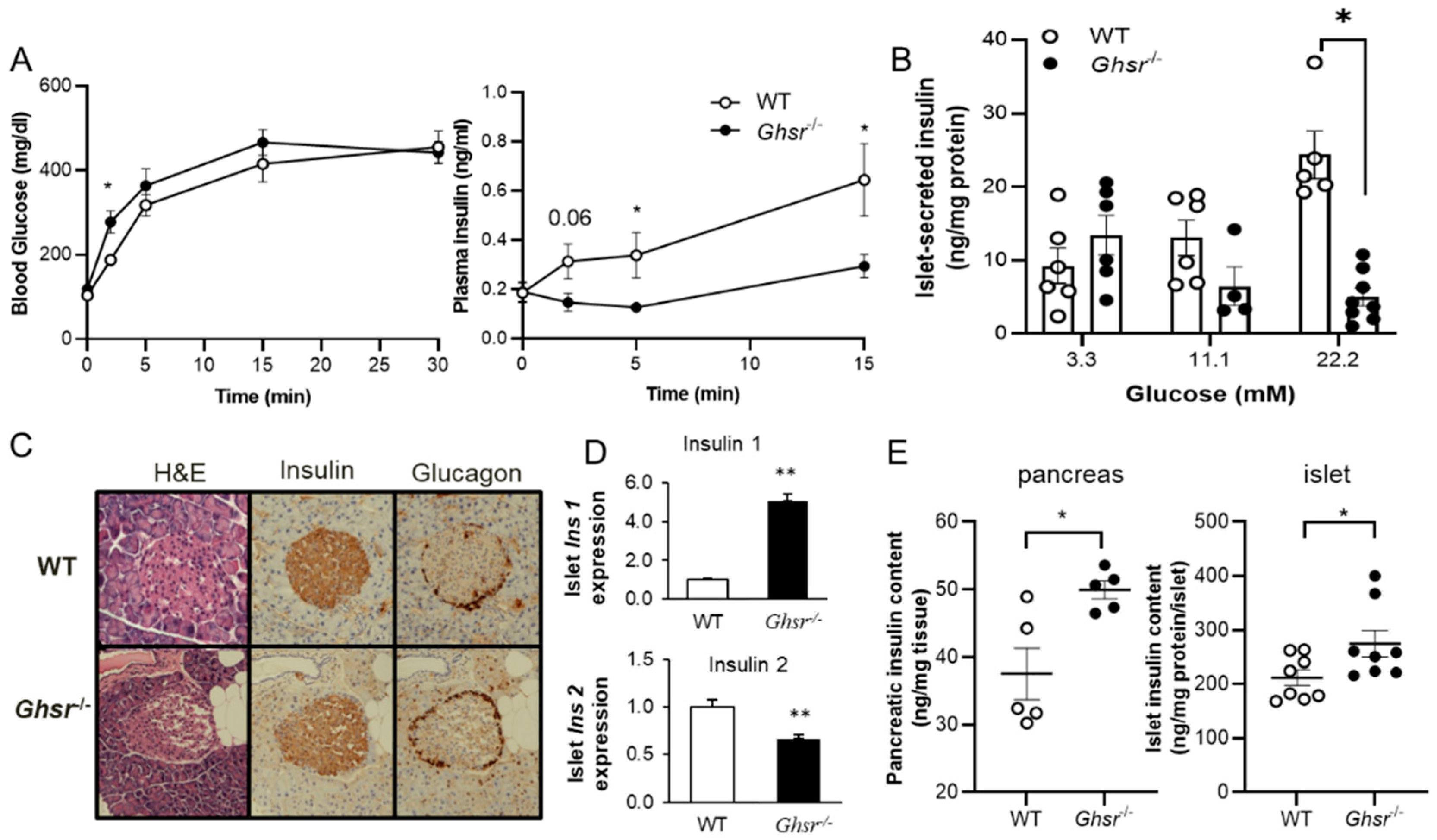
3.1. GHS-R Ablation Decreases Glucose-Stimulated Insulin Secretion (GSIS)
3.2. Ghsr−/− Islets Exhibit Lower ATP/ADP Ratio
3.3. Glucose Uptake Is Reduced in Ghsr-Ablated Islets
3.4. Identification of Molecular Mediators Governing GHS-R Induced GSIS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a Novel Growth Hormone-Releasing Acylated Peptide, Is Synthesized in a Distinct Endocrine Cell Type in the Gastrointestinal Tracts of Rats and Humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.L.; Pugh-Bernard, A.E.; Elghazi, L.; Sosa-Pineda, B.; Sussel, L. Ghrelin cells replace insulin-producing cells in two mouse models of pancreas development. Proc. Natl. Acad. Sci. USA 2004, 101, 2924–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, R.; Settanni, F.; Gallo, D.; Trovato, L.; Biancone, L.; Cantaluppi, V.; Nano, R.; Annunziata, M.; Campiglia, P.; Arnoletti, E.; et al. Obestatin Promotes Survival of Pancreatic Beta-Cells and Human Islets and Induces Expression of Genes Involved in the Regulation of -Cell Mass and Function. Diabetes 2007, 57, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Volante, M.; Allìa, E.; Gugliotta, P.; Funaro, A.; Broglio, F.; Deghenghi, R.; Muccioli, G.; Ghigo, E.; Papotti, M. Expression of Ghrelin and of the GH Secretagogue Receptor by Pancreatic Islet Cells and Related Endocrine Tumors. J. Clin. Endocrinol. Metab. 2002, 87, 1300–1308. [Google Scholar] [CrossRef]
- Wierup, N.; Yang, S.; McEvilly, R.J.; Mulder, H.; Sundler, F. Ghrelin Is Expressed in a Novel Endocrine Cell Type in Developing Rat Islets and Inhibits Insulin Secretion from INS-1 (832/13) Cells. J. Histochem. Cytochem. 2004, 52, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Date, Y.; Nakazato, M.; Hashiguchi, S.; Dezaki, K.; Mondal, M.S.; Hosoda, H.; Kojima, M.; Kangawa, K.; Arima, T.; Matsuo, H.; et al. Ghrelin Is Present in Pancreatic α-Cells of Humans and Rats and Stimulates Insulin Secretion. Diabetes 2002, 51, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, H.; Kangawa, K. The autonomic nervous system regulates gastric ghrelin secretion in rats. Regul. Pept. 2008, 146, 12–18. [Google Scholar] [CrossRef]
- Dezaki, K.; Hosoda, H.; Kakei, M.; Hashiguchi, S.; Watanabe, M.; Kangawa, K.; Yada, T. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: Implication in the glycemic control in rodents. Diabetes 2004, 53, 3142–3151. [Google Scholar] [CrossRef] [Green Version]
- Reimer, M.K.; Pacini, G.; Ahrén, B. Dose-Dependent Inhibition by Ghrelin of Insulin Secretion in the Mouse. Endocrinology 2003, 144, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, P.; Zheng, H.; Smith, R.G. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 4679–4684. [Google Scholar] [CrossRef] [Green Version]
- DiGruccio, M.R.; Mawla, A.M.; Donaldson, C.J.; Noguchi, G.M.; Vaughan, J.; Cowing-Zitron, C.; van der Meulen, T.; Huising, M.O. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 2016, 5, 449–458. [Google Scholar] [CrossRef]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988. [Google Scholar] [CrossRef]
- Granata, R.; Baragli, A.; Settanni, F.; Scarlatti, F.; Ghigo, E. Unraveling the role of the ghrelin gene peptides in the endocrine pancreas. J. Mol. Endocrinol. 2010, 45, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Anderwald-Stadler, M.; Krebs, M.; Promintzer, M.; Mandl, M.; Bischof, M.G.; Nowotny, P.; Kästenbauer, T.; Luger, A.; Prager, R.; Anderwald, C. Plasma obestatin is lower at fasting and not suppressed by insulin in insulin-resistant humans. Am. J. Physiol. Metab. 2007, 293, E1393–E1398. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Dowsett, G.K.C.; Mani, B.K.; Shankar, K.; Osborne-Lawrence, S.; Metzger, N.P.; Lam, B.Y.H.; Yeo, G.S.H.; Zigman, J.M. High Coexpression of the Ghrelin and LEAP2 Receptor GHSR With Pancreatic Polypeptide in Mouse and Human Islets. Endocrinology 2021, 162, bqab148. [Google Scholar] [CrossRef]
- Yu, X.-X.; Qiu, W.-L.; Yang, L.; Wang, Y.-C.; He, M.-Y.; Wang, D.; Zhang, Y.; Li, L.-C.; Zhang, J.; Wang, Y.; et al. Sequential progenitor states mark the generation of pancreatic endocrine lineages in mice and humans. Cell Res. 2021, 31, 886–903. [Google Scholar] [CrossRef]
- Dezaki, K.; Sone, H.; Koizumi, M.; Nakata, M.; Kakei, M.; Nagai, H.; Hosoda, H.; Kangawa, K.; Yada, T. Blockade of Pancreatic Islet–Derived Ghrelin Enhances Insulin Secretion to Prevent High-Fat Diet–Induced Glucose Intolerance. Diabetes 2006, 55, 3486–3493. [Google Scholar] [CrossRef] [Green Version]
- Chuang, J.-C.; Sakata, I.; Kohno, D.; Perello, M.; Osborne-Lawrence, S.; Repa, J.J.; Zigman, J.M. Ghrelin Directly Stimulates Glucagon Secretion from Pancreatic α-Cells. Mol. Endocrinol. 2011, 25, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.E.; Svendsen, B.Y.; Lam, B.; Yeo, G.S.H.; Holst, J.J.; Reimann, F.; Gribble, F.M. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 2016, 59, 2156–2165. [Google Scholar] [CrossRef] [Green Version]
- Dezaki, K.; Kakei, M.; Yada, T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: Novel signal transduction of ghrelin. Diabetes 2007, 56, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Jiang, H.; Zhang, H.; Smith, R.G. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc. Natl. Acad. Sci. USA 2012, 109, 19003–19008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurashina, T.; Dezaki, K.; Yoshida, M.; Rita, R.S.; Ito, K.; Taguchi, M.; Miura, R.; Tominaga, M.; Ishibashi, S.; Kakei, M.; et al. The β-cell GHSR and downstream cAMP/TRPM2 signaling account for insulinostatic and glycemic effects of ghrelin. Sci. Rep. 2015, 5, 14041. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Asnicar, M.; Saha, P.K.; Chan, L.; Smith, R.G. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metab. 2006, 3, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Saha, P.K.; Ma, X.; Henshaw, I.O.; Shao, L.; Chang, B.H.J.; Buras, E.D.; Tong, Q.; Chan, L.; McGuinness, O.P.; et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell 2011, 10, 996–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Lin, Y.; Lin, L.; Qin, G.; Pereira, F.A.; Haymond, M.W.; Butte, N.F.; Sun, Y. Ablation of ghrelin receptor in leptin-deficient ob/ob mice has paradoxical effects on glucose homeostasis when compared with ablation of ghrelin in ob/ob mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E422–E431. [Google Scholar] [CrossRef] [Green Version]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef]
- Aspinwall, C.A.; Lakey, J.R.T.; Kennedy, R.T. Insulin-stimulated Insulin Secretion in Single Pancreatic Beta Cells. J. Biol. Chem. 1999, 274, 6360–6365. [Google Scholar] [CrossRef] [Green Version]
- Assmann, A.; Hinault, C.; Kulkarni, R.N. Growth factor control of pancreatic islet regeneration and function. Pediatr. Diabetes 2009, 10, 14–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouche, C.; Lopez, X.; Fleischman, A.; Cypess, A.M.; O’Shea, S.; Stefanovski, D.; Bergman, R.N.; Rogatsky, E.; Stein, D.T.; Kahn, C.R.; et al. Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proc. Natl. Acad. Sci. USA 2010, 107, 4770–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halperin, F.; Lopez, X.; Manning, R.; Kahn, C.R.; Kulkarni, R.N.; Goldfine, A.B. Insulin Augmentation of Glucose-Stimulated Insulin Secretion Is Impaired in Insulin-Resistant Humans. Diabetes 2012, 61, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderwald, C.; Tura, A.; Grassi, A.; Krebs, M.; Szendroedi, J.; Roden, M.; Bischof, M.G.; Luger, A.; Pacini, G. Insulin Infusion During Normoglycemia Modulates Insulin Secretion According to Whole-Body Insulin Sensitivity. Diabetes Care 2011, 34, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.; Kahn, C. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic β Cells Creates an Insulin Secretory Defect Similar to that in Type 2 Diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Winnay, J.N.; Daniels, M.; Brüning, J.C.; Flier, S.N.; Hanahan, D.; Kahn, C.R. Altered function of insulin receptor substrate-1–deficient mouse islets and cultured β-cell lines. J. Clin. Investig. 1999, 104, R69–R75. [Google Scholar] [CrossRef] [Green Version]
- Withers, D.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.-M.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin Resistance and a Diabetes Mellitus-Like Syndrome in Mice Lacking the Protein Kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Kaneko, K.; Ueki, K.; Takahashi, N.; Hashimoto, S.; Okamoto, M.; Awazawa, M.; Okazaki, Y.; Ohsugi, M.; Inabe, K.; Umehara, T.; et al. Class IA Phosphatidylinositol 3-Kinase in Pancreatic β Cells Controls Insulin Secretion by Multiple Mechanisms. Cell Metab. 2010, 12, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Guillam, M.T.; Hummler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Deriaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Poykko, S.M.; Kellokoski, E.; Horkko, S.; Kauma, H.; Kesaniemi, Y.A.; Ukkola, O. Low Plasma Ghrelin Is Associated With Insulin Resistance, Hypertension, and the Prevalence of Type 2 Diabetes. Diabetes 2003, 52, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Purnell, J.Q.; Weigle, D.S.; Breen, P.; Cummings, D.E. Ghrelin Levels Correlate with Insulin Levels, Insulin Resistance, and High-Density Lipoprotein Cholesterol, But Not with Gender, Menopausal Status, or Cortisol Levels in Humans. J. Clin. Endocrinol. Metab. 2003, 88, 5747–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikezaki, A.; Hosoda, H.; Ito, K.; Iwama, S.; Miura, N.; Matsuoka, H.; Kondo, C.; Kojima, M.; Kangawa, K.; Sugihara, S. Fasting Plasma Ghrelin Levels Are Negatively Correlated With Insulin Resistance and PAI-1, but Not With Leptin, in Obese Children and Adolescents. Diabetes 2002, 51, 3408–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Yoon, C.Y.; Jang, P.G.; Park, Y.J.; Shin, C.S.; Park, H.S.; Ryu, J.W.; Pak, Y.K.; Park, J.Y.; Lee, K.U.; et al. The Mitogenic and Antiapoptotic Actions of Ghrelin in 3T3-L1 Adipocytes. Mol. Endocrinol. 2004, 18, 2291–2301. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, Y.; Zhang, W. The Growth Hormone Secretagogue Receptor: Its Intracellular Signaling and Regulation. Int. J. Mol. Sci. 2014, 15, 4837–4855. [Google Scholar] [CrossRef]
- Murata, M.; Okimura, Y.; Iida, K.; Matsumoto, M.; Sowa, H.; Kaji, H.; Kojima, M.; Kangawa, K.; Chihara, K. Ghrelin Modulates the Downstream Molecules of Insulin Signaling in Hepatoma Cells. J. Biol. Chem. 2002, 277, 5667–5674. [Google Scholar] [CrossRef] [Green Version]
- Yeung, E.; Zhang, C.; Mumford, S.; Ye, A.; Trevisan, M.; Chen, L.; Browne, R.W.; Wactawski-Wende, J.; Schisterman, E. Longitudinal Study of Insulin Resistance and Sex Hormones over the Menstrual Cycle: The BioCycle Study. J. Clin. Endocrinol. Metab. 2010, 95, 5435–5442. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-S.; Yuan, Y.-H.; Tu, H.-J.; Liang, Q.-L.; Dai, L.-J. A protocol for islet isolation from mouse pancreas. Nat. Protoc. 2009, 4, 1649–1652. [Google Scholar] [CrossRef]
- Villarreal, D.; Pradhan, G.; Wu, C.S.; Allred, C.D.; Guo, S.; Sun, Y. A Simple High Efficiency Protocol for Pancreatic Islet Isolation from Mice. J. Vis. Exp. 2019, 150, e57048. [Google Scholar] [CrossRef]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.; Kopchick, J.J.; Liu, J.-L. Growth hormone receptor gene deficiency causes delayed insulin responsiveness in skeletal muscles without affecting compensatory islet cell overgrowth in obese mice. Am. J. Physiol. Metab. 2006, 291, E491–E498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Lin, L.; Qin, G.; Lu, X.; Fiorotto, M.; Dixit, V.D.; Sun, Y. Ablations of Ghrelin and Ghrelin Receptor Exhibit Differential Metabolic Phenotypes and Thermogenic Capacity during Aging. PLoS ONE 2011, 6, e16391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Sun, Y.; Butte, N.F.; Garcia, J.M.; Smith, R.G. Characterization of Adult Ghrelin and Ghrelin Receptor Knockout Mice under Positive and Negative Energy Balance. Endocrinology 2007, 149, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Wentworth, B.M.; Schaefer, I.M.; Villa-Komaroff, L.; Chirgwin, J.M. Characterization of the two nonallelic genes encoding mouse preproinsulin. J. Mol. Evol. 1986, 23, 305–312. [Google Scholar] [CrossRef]
- Shiao, M.-S.; Liao, B.-Y.; Long, M.; Yu, H.-T. Adaptive Evolution of the Insulin Two-Gene System in Mouse. Genet. 2008, 178, 1683–1691. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Gao, L.; Wang, K.; Ma, X.; Chang, X.; Shi, J.-H.; Zhang, Y.; Yin, K.; Liu, Z.; Shi, Y.; et al. Knockin of Cre Gene at Ins2 Locus Reveals No Cre Activity in Mouse Hypothalamic Neurons. Sci. Rep. 2016, 6, 20438. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Almaça, J.; Dadi, P.K.; Hong, H.; Sakamoto, W.; Rossi, M.; Lee, R.; Vierra, N.C.; Lu, H.; Cui, Y.; et al. β-arrestin-2 is an essential regulator of pancreatic β-cell function under physiological and pathophysiological conditions. Nat. Commun. 2017, 8, 14295. [Google Scholar] [CrossRef]
- Brüning, D.; Reckers, K.; Drain, P.; Rustenbeck, I. Glucose but not KCl diminishes submembrane granule turnover in mouse beta-cells. J. Mol. Endocrinol. 2017, 59, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and Metabolic Evidence for Mitochondrial Defects Associated with β-Cell Dysfunction in a Mouse Model of Type 2 Diabetes. Diabetes 2009, 59, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2014, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gu, W.; Chen, C. Knocking down Insulin Receptor in Pancreatic Beta Cell lines with Lentiviral-Small Hairpin RNA Reduces Glucose-Stimulated Insulin Secretion via Decreasing the Gene Expression of Insulin, GLUT2 and Pdx1. Int. J. Mol. Sci. 2018, 19, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watada, H.; Kajimoto, Y.; Miyagawa, J.-I.; Hanafusa, T.; Hamaguchi, K.; Matsuoka, T.-A.; Yamamoto, K.; Matsuzawa, Y.; Kawamori, R.; Yamasaki, Y. PDX-1 Induces Insulin and Glucokinase Gene Expressions in αTC1 Clone 6 Cells in the Presence of Betacellulin. Diabetes 1996, 45, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Maechler, P.; Ritz-Laser, B.; Hagenfeldt, K.A.; Ishihara, H.; Philippe, J.; Wollheim, C.B. Pdx1 Level Defines Pancreatic Gene Expression Pattern and Cell Lineage Differentiation. J. Biol. Chem. 2001, 276, 25279–25286. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2006, 50, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.-A.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA Regulates Expression of Genes Important to Islet β-Cell Function. Mol. Endocrinol. 2007, 21, 2764–2774. [Google Scholar] [CrossRef]
- Pradhan, G.; Wu, C.S.; Lee, J.H.; Kanikarla, P.; Guo, S.; Yechoor, V.K.; Samson, S.L.; Sun, Y. Obestatin stimulates glucose-induced insulin secretion through ghrelin receptor GHS-R. Sci. Rep. 2017, 7, 979. [Google Scholar] [CrossRef]
- Wu, C.-S.; Bongmba, O.Y.N.; Lee, J.H.; Tuchaai, E.; Zhou, Y.; Li, D.-P.; Xue, B.; Chen, Z.; Sun, Y. Ghrelin receptor in agouti-related peptide neurones regulates metabolic adaptation to calorie restriction. J. Neuroendocr. 2019, 31, e12763. [Google Scholar] [CrossRef] [PubMed]
- Zigman, J.M.; Nakano, Y.; Coppari, R.; Balthasar, N.; Marcus, J.N.; Lee, C.E.; Jones, J.E.; Deysher, A.E.; Waxman, A.R.; White, R.D.; et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J. Clin. Investig. 2005, 115, 3564–3572. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Wu, C.-S.; Villarreal, D.; Lee, J.; Han, H.; Gaharwar, A.; Tian, Y.; Fu, W.; Guo, S.; Smith, R.; et al. β Cell GHS-R Regulates Insulin Secretion and Sensitivity. Int. J. Mol. Sci. 2021, 22, 3950. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Niu, J.; Zhang, A.; Svendsen, B.; Campbell, J.E.; D’Alessio, D.A.; Tong, J. Intraislet Ghrelin Signaling Does Not Regulate Insulin Secretion From Adult Mice. Diabetes 2019, 68, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Leroux, L.; Desbois, P.; Lamotte, L.; Duvillié, B.; Cordonnier, N.; Jackerott, M.; Jami, J.; Bucchini, D.; Joshi, R.L. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 2001, 50 (Suppl. 1), 150–153. [Google Scholar] [CrossRef] [Green Version]
- Roderigo-Milne, H.; Hauge-Evans, A.C.; Persaud, S.; Jones, P. Differential expression of insulin genes 1 and 2 in MIN6 cells and pseudoislets. Biochem. Biophys. Res. Commun. 2002, 296, 589–595. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; Van De Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Wang, W.; Upshaw, L.; Strong, D.M.; Robertson, R.P.; Reems, J. Increased oxygen consumption rates in response to high glucose detected by a novel oxygen biosensor system in non-human primate and human islets. J. Endocrinol. 2005, 185, 445–455. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Takamatsu, S.; Minowa, M.T.; Yoshida, A.; Takeuchi, M.; Marth, J.D. Dietary and Genetic Control of Glucose Transporter 2 Glycosylation Promotes Insulin Secretion in Suppressing Diabetes. Cell 2005, 123, 1307–1321. [Google Scholar] [CrossRef] [Green Version]
- Raum, J.C.; Gerrish, K.; Artner, I.; Henderson, E.; Guo, M.; Sussel, L.; Schisler, J.; Newgard, C.B.; Stein, R. FoxA2, Nkx2.2, and PDX-1 Regulate Islet β-Cell-Specific mafA Expression through Conserved Sequences Located between Base Pairs −8118 and −7750 Upstream from the Transcription Start Site. Mol. Cell. Biol. 2006, 26, 5735–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Sharma, A.; Zangen, D.H.; Bonner-Weir, S. Transcription factor abnormalities as a cause of beta cell dysfunction in diabetes: A hypothesis. Acta Diabetol. 1997, 34, 177–184. [Google Scholar] [CrossRef]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. β-Cell-specific inactivation of the mouseIpf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, T.-A.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.-A.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the Large Maf Transcription Family Regulate Insulin Gene Transcription in Islet β Cells. Mol. Cell. Biol. 2003, 23, 6049–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaras, S.E.; Zhao, L.; Means, A.; Henderson, E.; Matsuoka, T.-A.; Stein, R. The Islet beta Cell-enriched RIPE3b1/Maf Transcription Factor Regulates pdx-1 Expression. J. Biol. Chem. 2003, 278, 12263–12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunton, J.E.; Kulkarni, R.N.; Yim, S.; Okada, T.; Hawthorne, W.J.; Tseng, Y.-H.; Roberson, R.S.; Ricordi, C.; O’Connell, P.J.; Gonzalez, F.J.; et al. Loss of ARNT/HIF1β Mediates Altered Gene Expression and Pancreatic-Islet Dysfunction in Human Type 2 Diabetes. Cell 2005, 122, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Norquay, L.D.; D’Aquino, K.E.; Opare-Addo, L.M.; Kuznetsova, A.; Haas, M.; Bluestone, J.A.; White, M.F. Insulin Receptor Substrate-2 in β-Cells Decreases Diabetes in Nonobese Diabetic Mice. Endocrinology 2009, 150, 4531–4540. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Bernal-Mizrachi, E.; Alejandro, E.; Han, Z.; Kalynyak, T.B.; Li, H.; Beith, J.L.; Gross, J.; Warnock, G.L.; Townsend, R.R.; et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 19575–19580. [Google Scholar] [CrossRef] [Green Version]
- Escribano, Ó.; Gomez-Hernandez, A.; Diaz-Castroverde, S.; Nevado, C.; García, G.; Otero, Y.F.; Perdomo, L.; Beneit, N.; Benito, M. Insulin receptor isoform A confers a higher proliferative capability to pancreatic beta cells enabling glucose availability and IGF-I signaling. Mol. Cell. Endocrinol. 2015, 409, 82–91. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.D.; Dula, S.B.; Corbin, K.L.; Wu, R.; Nunemaker, C.S. A Practical Guide to Rodent Islet Isolation and Assessment. Biol. Proced. Online 2009, 11, 3–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardooz, H.; Asl, S.Z.; Naseri, M.G. Effect of chronic psychological stress on insulin release from rat isolated pancreatic islets. Life Sci. 2006, 79, 57–62. [Google Scholar] [CrossRef]
- Zardooz, H.; Zahediasl, S.; Rostamkhani, F.; Farrokhi, B.; Nasiraei, S.; Kazeminezhad, B.; Gholampour, R. Effects of acute and chronic psychological stress on isolated islets’ insulin release. EXCLI J. 2012, 11, 163–175. [Google Scholar]
- Gray, S.M.; Page, L.C.; Tong, J. Ghrelin regulation of glucose metabolism. J. Neuroendocr. 2019, 31, e12705. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Fiori, J.L.; Shin, Y.-K.; Okun, E.; Kim, J.S.; Rapp, P.R.; Egan, J.M. Pancreatic polypeptide inhibits somatostatin secretion. FEBS Lett. 2014, 588, 3233–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Lee, Y.L.; Kwon, H.Y. Effects of pancreatic polypeptide on insulin action in exocrine secretion of isolated rat pancreas. J. Physiol. 1993, 463, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Egido, E.M.; Rodriguez-Gallardo, J.; Silvestre, R.A.; Marco, J. Inhibitory effect of ghrelin on insulin and pancreatic somatostatin secretion. Eur. J. Endocrinol. 2002, 146, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Mani, B.K.; Shankar, K.; Zigman, J.M. Ghrelin’s Relationship to Blood Glucose. Endocrinology 2019, 160, 1247–1261. [Google Scholar] [CrossRef]
- Wren, A.M.; Small, C.J.; Ward, H.L.; Murphy, K.G.; Dakin, C.L.; Taheri, S.; Kennedy, A.R.; Roberts, G.H.; Morgan, D.G.; Ghatei, M.A.; et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 2020, 14, 4325–4328. [Google Scholar] [CrossRef]
- Arosio, M.; Ronchi, C.L.; Gebbia, C.; Cappiello, V.; Beck-Peccoz, P.; Peracchi, M. Stimulatory Effects of Ghrelin on Circulating Somatostatin and Pancreatic Polypeptide Levels. J. Clin. Endocrinol. Metab. 2003, 88, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.C.; Bauchle, C.J.; Rouault, A.A.; Stephens, S.B.; Sebag, J.A. The Insulinostatic Effect of Ghrelin Requires MRAP2 Expression in δ Cells. Iscience 2020, 23, 101216. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, N.; Gill, D.A.S.; Davies, R.; Loveridge, N.; Houston, P.A.; Robinson, I.C.A.F.; Wells, T. Ghrelin and Des-Octanoyl Ghrelin Promote Adipogenesis Directly in Vivo by a Mechanism Independent of the Type 1a Growth Hormone Secretagogue Receptor. Endocrinology 2004, 145, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Delhanty, P.J.D.; van der Eerden, B.; Van Der Velde, M.; Gauna, C.; Pols, H.A.P.; Jahr, H.; Chiba, H.; Van Der Lely, A.J.; Van Leeuwen, J.P.T.M. Ghrelin and unacylated ghrelin stimulate human osteoblast growth via mitogen-activated protein kinase (MAPK)/phosphoinositide 3-kinase (PI3K) pathways in the absence of GHS-R1a. J. Endocrinol. 2006, 188, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meulen, T.; Donaldson, C.J.; Caceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Shankar, K.; Takemi, S.; Gupta, D.; Varshney, S.; Mani, B.K.; Osborne-Lawrence, S.; Metzger, N.P.; Richard, C.P.; Berglund, E.D.; Zigman, J.M. Ghrelin cell-expressed insulin receptors mediate meal- and obesity-induced declines in plasma ghrelin. JCI Insight 2021, 6, e146983. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradhan, G.; Lee, J.H.; Wu, C.-S.; Wang, H.; Lin, L.; Donti, T.; Graham, B.H.; Rajan, A.S.; Balasubramanyam, A.; Samson, S.L.; et al. Mechanistic Investigation of GHS-R Mediated Glucose-Stimulated Insulin Secretion in Pancreatic Islets. Biomolecules 2022, 12, 407. https://doi.org/10.3390/biom12030407
Pradhan G, Lee JH, Wu C-S, Wang H, Lin L, Donti T, Graham BH, Rajan AS, Balasubramanyam A, Samson SL, et al. Mechanistic Investigation of GHS-R Mediated Glucose-Stimulated Insulin Secretion in Pancreatic Islets. Biomolecules. 2022; 12(3):407. https://doi.org/10.3390/biom12030407
Chicago/Turabian StylePradhan, Geetali, Jong Han Lee, Chia-Shan Wu, Hongying Wang, Ligen Lin, Taraka Donti, Brett H. Graham, Arun S. Rajan, Ashok Balasubramanyam, Susan L. Samson, and et al. 2022. "Mechanistic Investigation of GHS-R Mediated Glucose-Stimulated Insulin Secretion in Pancreatic Islets" Biomolecules 12, no. 3: 407. https://doi.org/10.3390/biom12030407
APA StylePradhan, G., Lee, J. H., Wu, C. -S., Wang, H., Lin, L., Donti, T., Graham, B. H., Rajan, A. S., Balasubramanyam, A., Samson, S. L., Guo, S., & Sun, Y. (2022). Mechanistic Investigation of GHS-R Mediated Glucose-Stimulated Insulin Secretion in Pancreatic Islets. Biomolecules, 12(3), 407. https://doi.org/10.3390/biom12030407