
Evidence of Failed Resolution Mechanisms in Arrhythmogenic Inflammation, Fibrosis and Right Heart Disease


Abstract
:1. Introduction
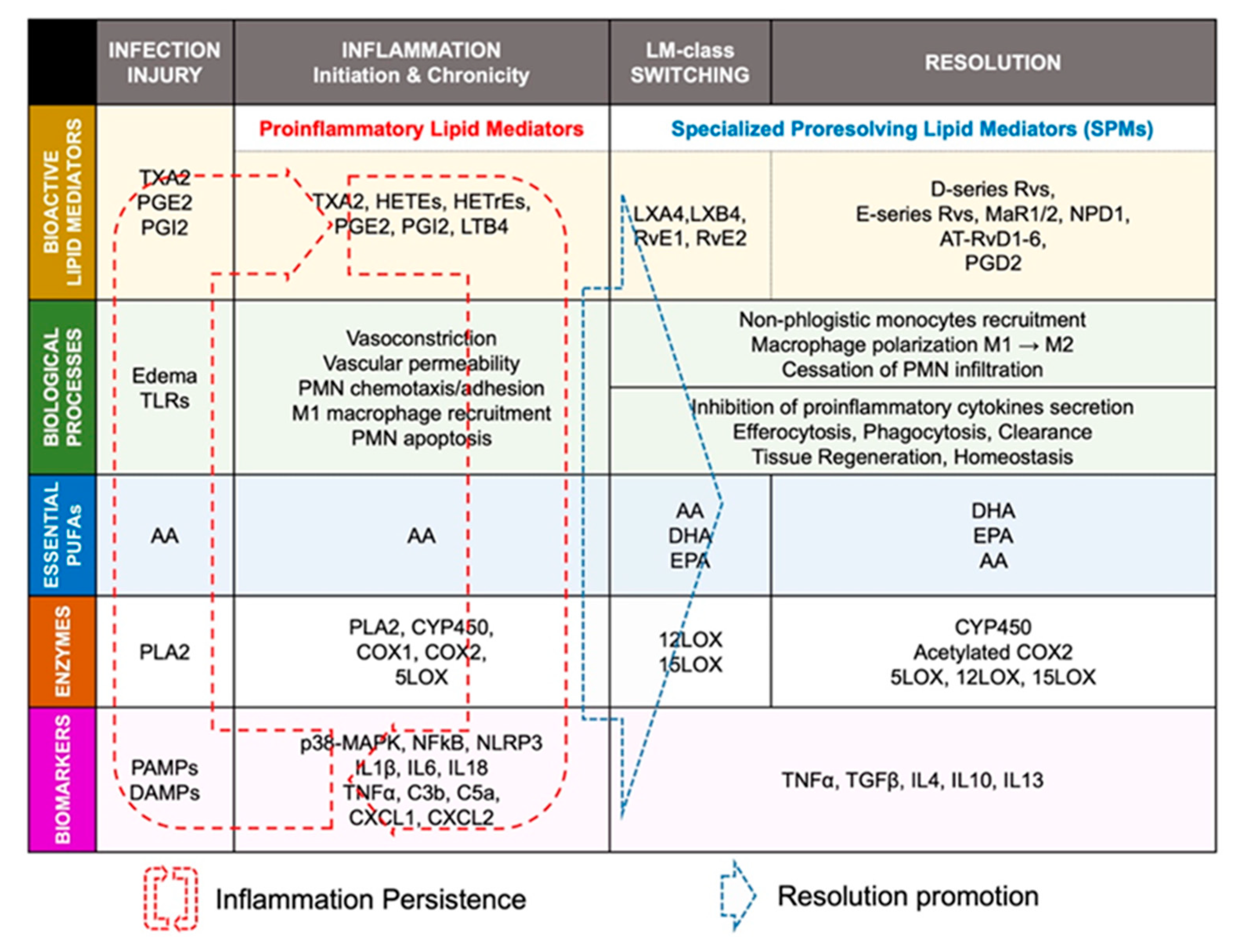
2. Biomolecular Paradigm of Active Resolution Mechanisms in the Heart
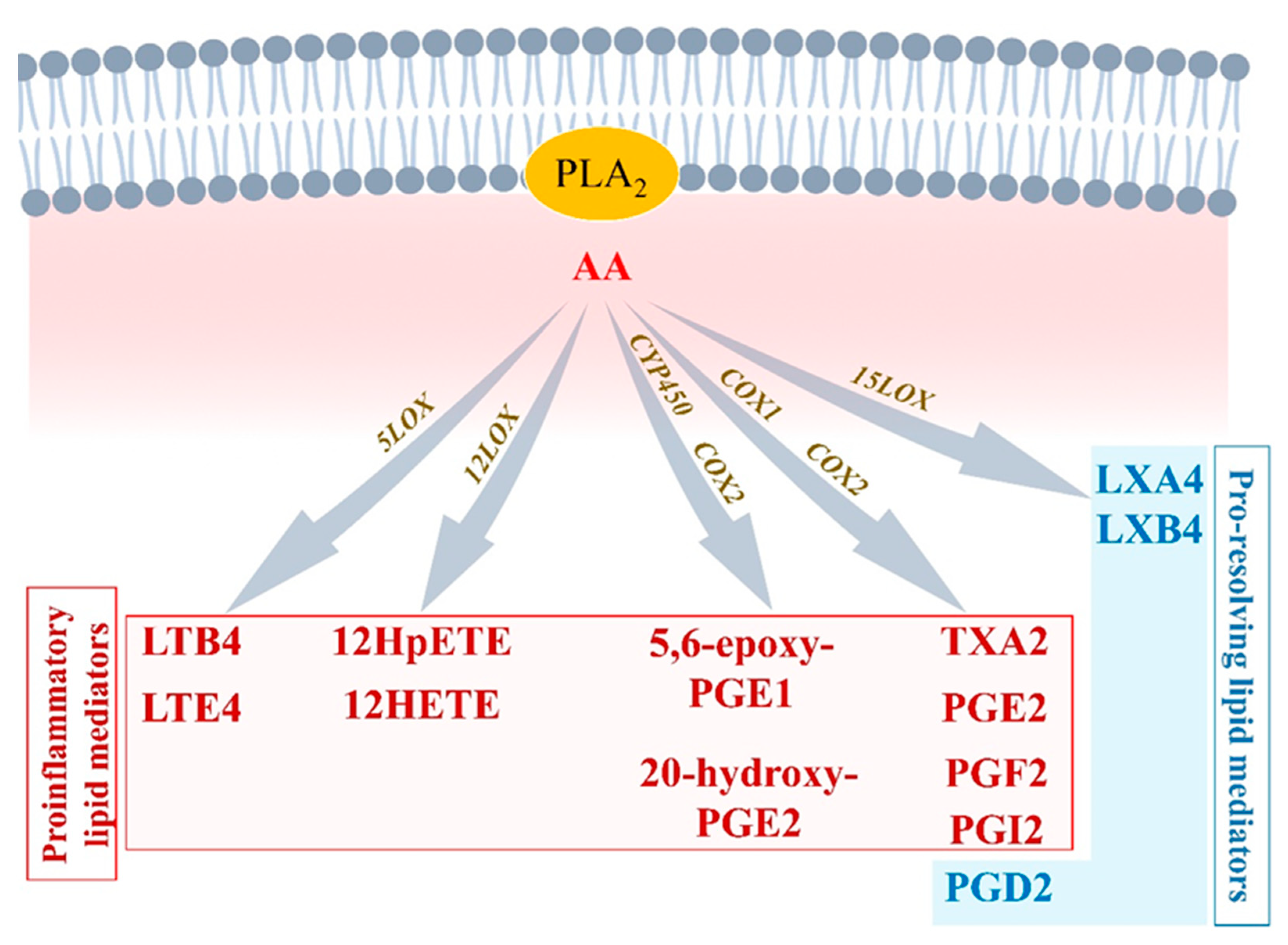
2.1. Initiation Phase of Inflammation: Central Regulatory Role of Arachidonic Acid
2.1.1. Arachidonic Acid Metabolism by Cytochrome P450
2.1.2. Arachidonic Acid Metabolism by COX1 and COX2
2.1.3. Arachidonic Acid Metabolism by 5-LOX
2.2. Lipid-Mediator Class Switching: Transition from Pro-Inflammatory to Pro-Resolution Signals
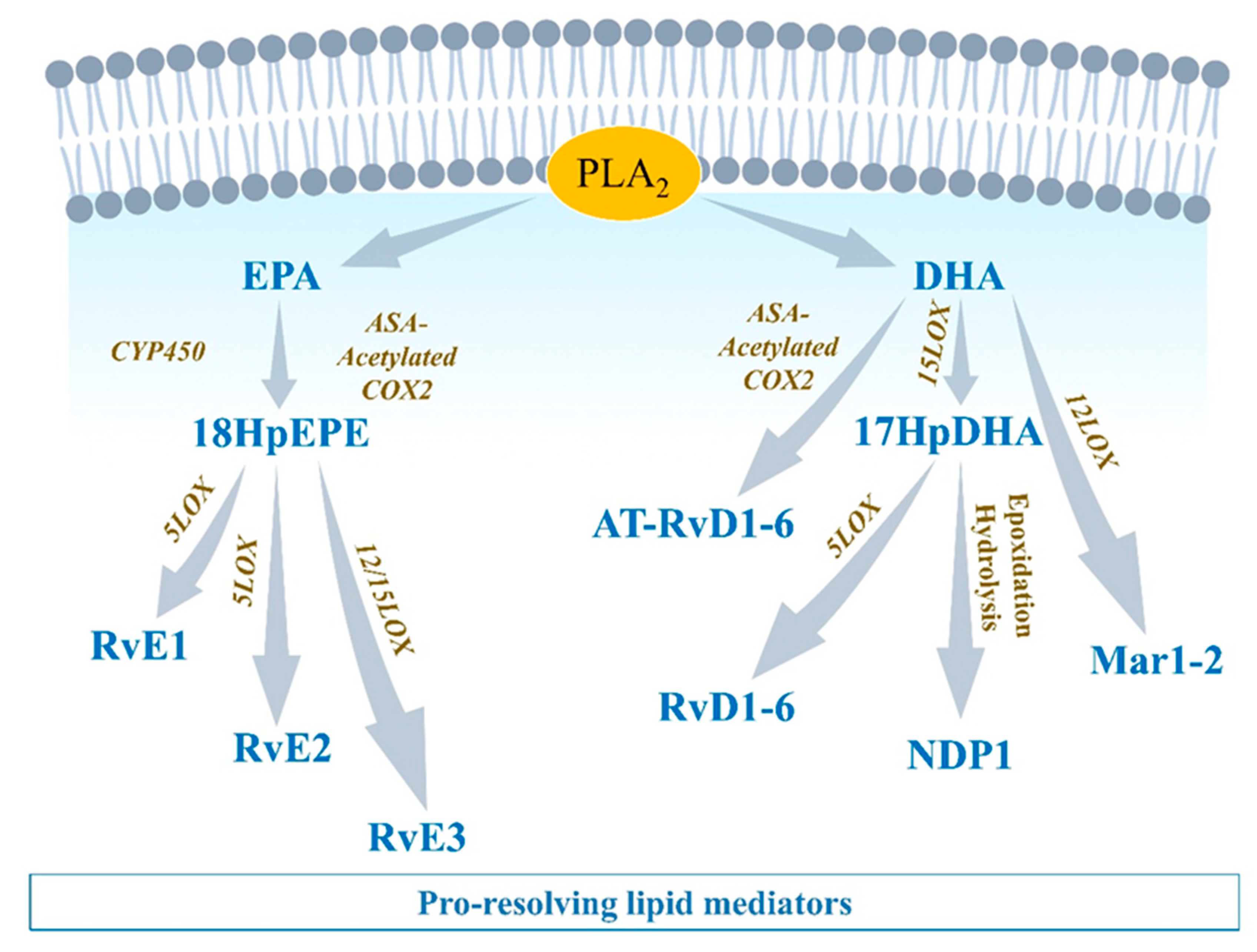
2.3. Resolution of Inflammation: SPMs-Mediated Efferocytosis and Homeostasis
2.3.1. EPA-Derived Specialized Pro-Resolving Mediators
2.3.2. DHA-Derived Specialized Pro-Resolving Mediators
2.3.3. Arachidonic Acid-Derived Specialized Pro-Resolving Mediators
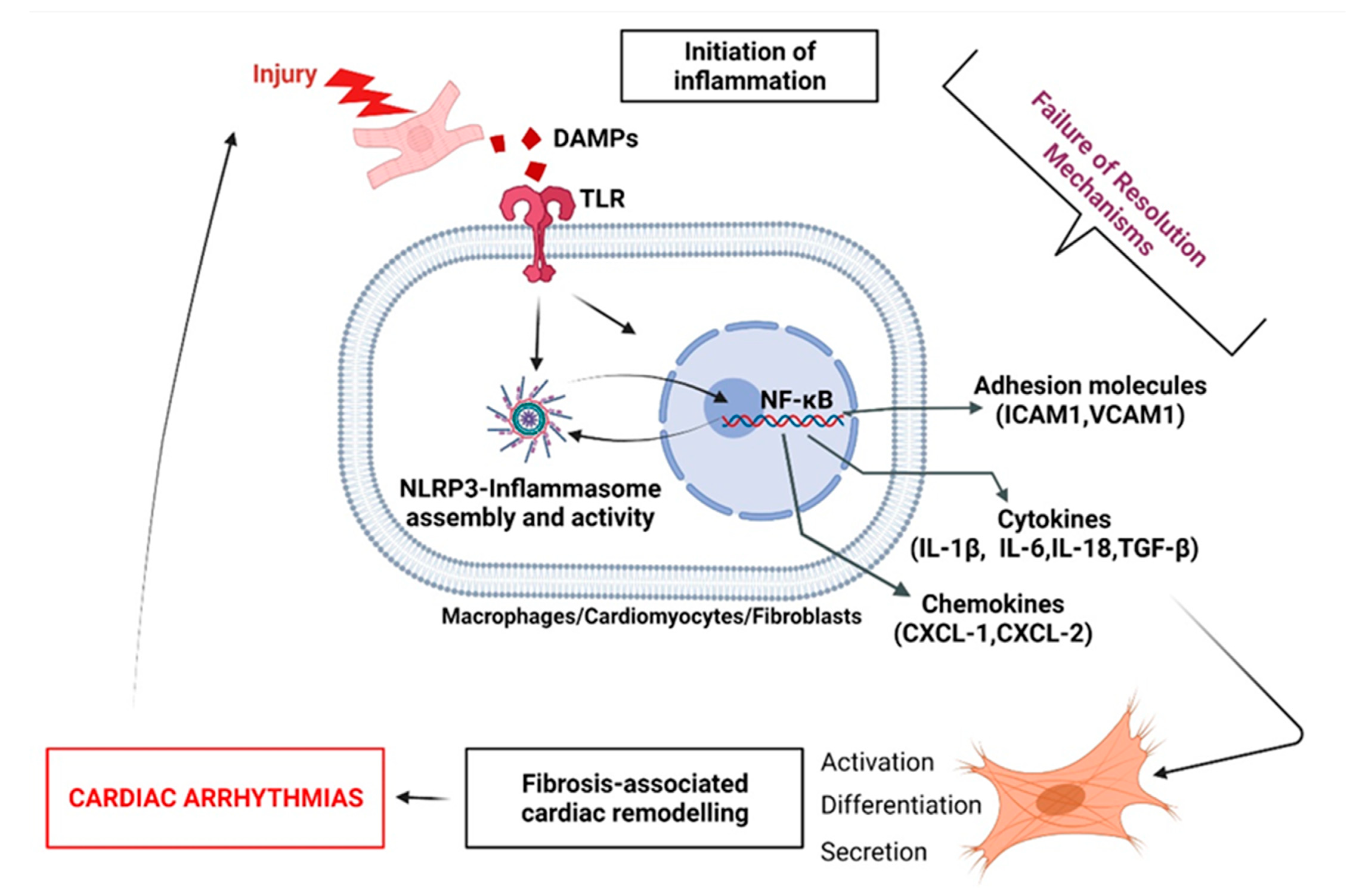
2.4. ‘Failed Resolution Mechanisms’ in the Development of Chronic Inflammation and Heart Diseases
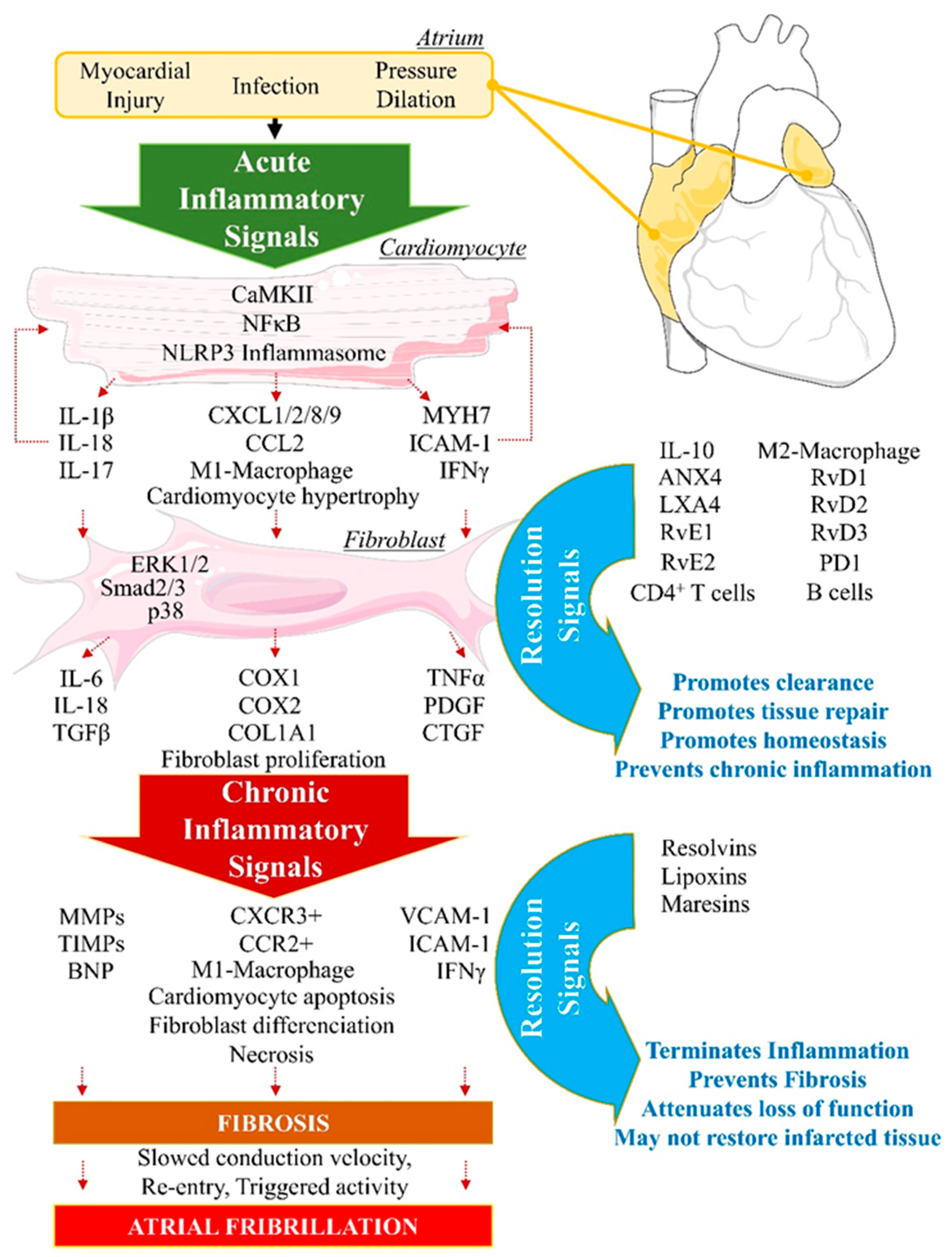
3. Description of ‘Failed Resolution Mechanisms’ in Cardiac Arrhythmogenic Remodeling
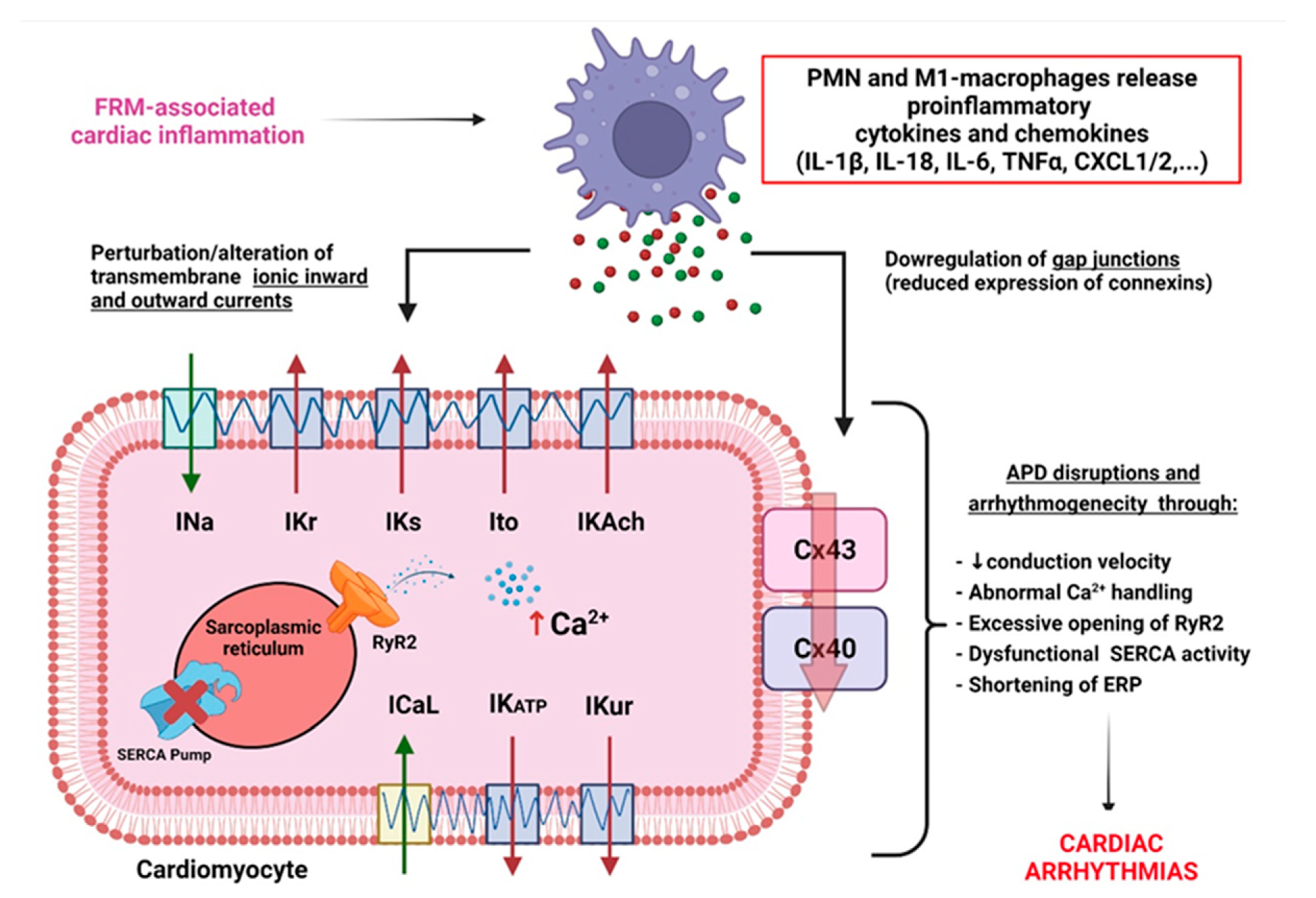
3.1. FRM Associated with Cardiac Electrical Conduction Abnormalities
3.2. FRM Associated with Cardiac ECM’s Arrhythmogenic Structural Remodeling
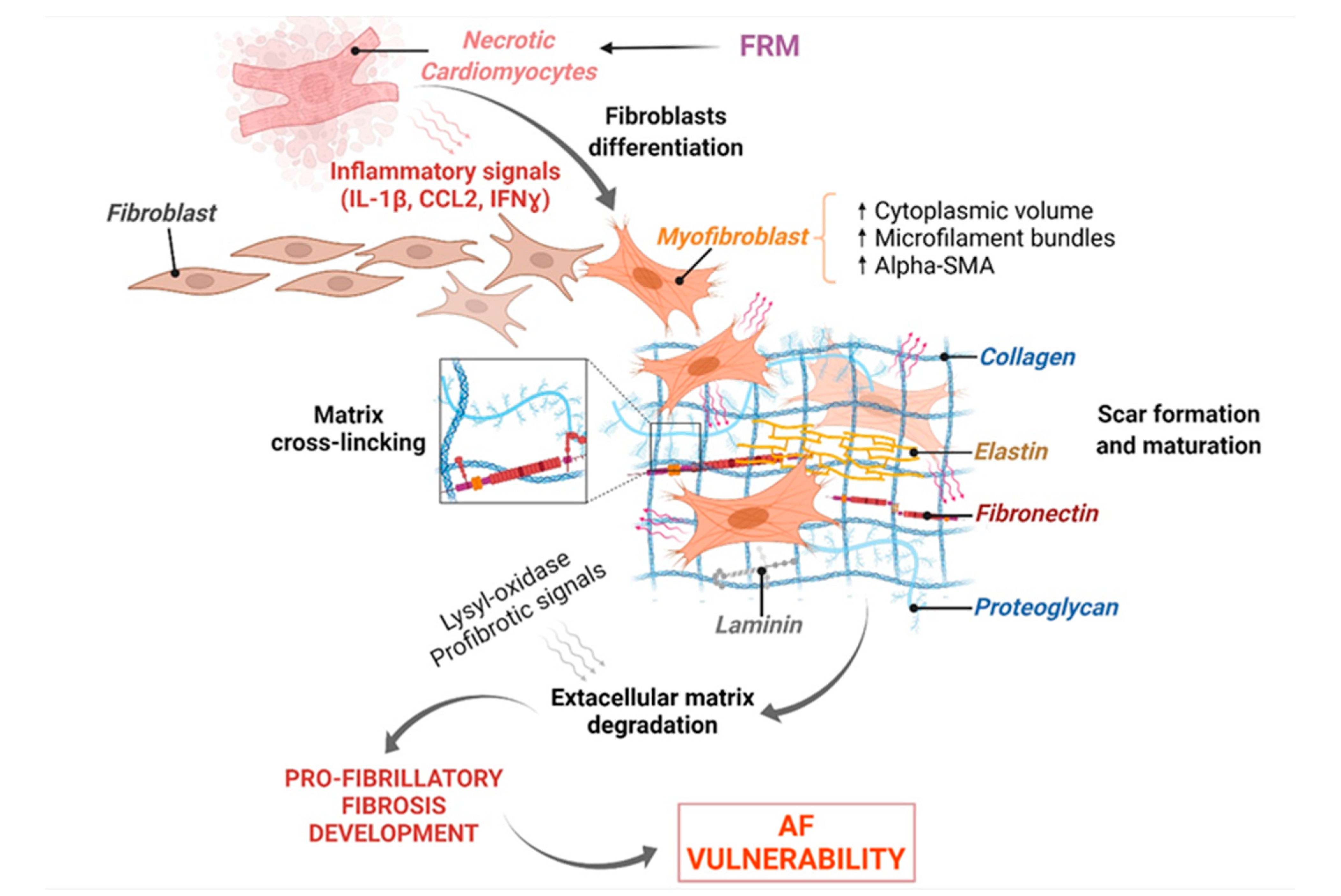
3.3. FRM Associated with Abnormal Cardiac Fibroblasts’ Remodeling and Atrial Fibrosis
3.3.1. Fibroblast Response to Inflammation Initiation
3.3.2. FB-Induced Expression of Key FRM-Promoting Biomarkers

4. Arrhythmogenic FRM in the Context of Right Heart Disease
4.1. Generalities on RHD-Induced Arrhythmogenicity
4.2. Resolution-Promoting Strategies in Monocrotaline and Sugen-Hypoxia Models of RHD and FRM Associated with Cardiac Arrhythmias
5. Conclusions
6. Highlights
- Initiation of inflammation is required to combat cardiac insults.
- Arrhythmogenic events may include inhibition of bio-molecularly active lipid-mediator class-switching and resolution.
- Future therapeutic strategies targeting cardiac inflammation must consider the complex equation of not inhibiting the required initiation-processes of inflammation while promoting resolution mechanisms.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Atrial Fibrillation |
| CM | Cardiomyocytes |
| ECM | Extracellular Matrix |
| FB | Cardiac Fibroblasts |
| FRM | Failed Resolution Mechanisms |
| IL | Interleukin |
| MI | Myocardial Infarction |
| RA | Right Atrium |
| RHD | Right Heart Disease |
| SPMs | Specialized Pro-Resolving Mediators |
References
- Andrade, J.; Khairy, P.; Dobrev, D.; Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 2014, 114, 1453–1468. [Google Scholar] [CrossRef]
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of Inflammation in Atrial Fibrillation Pathophysiology and Management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2013, 70, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, A.J.; Engler, A.J. Regenerative cross talk between cardiac cells and macrophages. Am. J. Physiol. Circ. Physiol. 2021, 320, H2211–H2221. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Scott, L., Jr.; Li, N.; Dobrev, D. Role of inflammatory signaling in atrial fibrillation. Int. J. Cardiol. 2019, 287, 195–200. [Google Scholar] [CrossRef]
- Hiram, R. Cardiac cytokine therapy? Relevance of targeting inflammatory mediators to combat cardiac arrhythmogenic remodeling. IJC Heart Vasc. 2021, 37, 100918. [Google Scholar] [CrossRef]
- Akdis, D.; Chen, K.; Saguner, A.M.; Stämpfli, S.F.; Chen, X.; Chen, L.; Rao, M.; Haegeli, L.M.; Tanner, F.C.; Brunckhorst, C.; et al. Clinical Characteristics of Patients with a Right Ventricular Thrombus in Arrhythmogenic Right Ventricular Cardiomyopathy. Thromb. Haemost. 2019, 119, 1373–1378. [Google Scholar] [CrossRef]
- Keramida, K.; Lazaros, G.; Nihoyannopoulos, P. Right ventricular involvement in hypertrophic cardiomyopathy: Patterns and implications. Hell. J. Cardiol. 2018, 61, 3–8. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Pencic, B.; Sljivic, A.; Ivanovic, B.; Neskovic, A.; Scepanovic, R.; Celic, V. High-normal blood pressure impacts the right heart mechanics: A three-dimensional echocardiography and two-dimensional speckle tracking imaging study. Blood Press. Monit. 2014, 19, 145–152. [Google Scholar] [CrossRef]
- Waligóra, M.; Tyrka, A.; Miszalski-Jamka, T.; Urbańczyk-Zawadzka, M.; Podolec, P.; Kopeć, G. Right atrium enlargement predicts clinically significant supraventricular arrhythmia in patients with pulmonary arterial hypertension. Heart Lung 2018, 47, 237–242. [Google Scholar] [CrossRef]
- Scheel, P.J., III; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134. [Google Scholar] [CrossRef]
- Sun, X.-Q.; Abbate, A.; Bogaard, H.-J. Role of cardiac inflammation in right ventricular failure. Cardiovasc. Res. 2017, 113, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285, Erratum in Nat. Rev. Cardiol. 2021, 18, 735. [Google Scholar] [CrossRef]
- Everett, T.H., IV; Olgin, J.E. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007, 4 (Suppl. 3), S24–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card. Electrophysiol. Clin. 2021, 13, 25–35. [Google Scholar] [CrossRef]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A (2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Sofogianni, A.; Alkagiet, S.; Tziomalos, K. Lipoprotein-associated Phospholipase A2 and Coronary Heart Disease. Curr. Pharm. Des. 2018, 24, 291–296. [Google Scholar] [CrossRef]
- Madjid, M.; Ali, M.; Willerson, J.T. Lipoprotein-associated phospholipase A2 as a novel risk marker for cardiovascular disease: A systematic review of the literature. Tex. Heart Inst. J. 2010, 37, 25–39. [Google Scholar]
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforinit, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A.; et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Zeldin, D. Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol. 2002, 13, 273–283. [Google Scholar] [CrossRef]
- Rossi, A.G.; O’Flaherty, J.T. Bioactions of 5-hydroxyicosatetraenoate and its interaction with platelet-activating factor. Lipids 1991, 26, 1184–1188. [Google Scholar] [CrossRef]
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic Acid Oxygenation by COX-1 and COX-2. J. Biol. Chem. 1999, 274, 22903–22906. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of regulation. J. Lipid Res. 2009, 50, S40–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maayah, Z.H.; El-Kadi, A.O.S. 5-, 12- and 15-Hydroxyeicosatetraenoic acids induce cellular hypertrophy in the human ventricular cardiomyocyte, RL-14 cell line, through MAPK- and NF-κB-dependent mechanism. Arch. Toxicol. 2015, 90, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Khan, H.; Xiao, J.; Cheang, W.S. Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 12029. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252. [Google Scholar] [CrossRef]
- Fiore, S.; Brezinski, M.E.; Sheppard, K.-A.; Serhan, C.N. The Lipoxin Biosynthetic Circuit and their Actions with Human Neutrophils. Adv. Exp. Med. Biol. 1991, 314, 109–132. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.; ASchmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Reina-Couto, M.; Carvalho, J.; Valente, M.J.; Vale, L.; Afonso, J.; Carvalho, F.; Bettencourt, P.; Sousa, T.; Albino-Teixeira, A. Impaired resolution of inflammation in human chronic heart failure. Eur. J. Clin. Investig. 2014, 44, 527–538. [Google Scholar] [CrossRef]
- Shi, Y.; Pan, H.; Zhang, H.-Z.; Zhao, X.-Y.; Jin, J.; Wang, H.-Y. Lipoxin A4 mitigates experimental autoimmune myocarditis by regulating inflammatory response, NF-κB and PI3K/Akt signaling pathway in mice. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1850–1859. [Google Scholar] [PubMed]
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 9999. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Yoshida, M.; Arita, M. Omega-3 fatty acid-derived mediators that control inflammation and tissue homeostasis. Int. Immunol. 2019, 31, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, J.A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: A current perspective on cardiovascular risks. Can. J. Cardiol. 2007, 23, 125–131. [Google Scholar] [CrossRef] [Green Version]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: An overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004, 73, 155–172. [Google Scholar] [CrossRef]
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2015, 785, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Alsaleh, N.; Lee, C.R.; Seubert, J.M. Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 2014, 74, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiù, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 2020, 74, 100894. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zhang, X.; Yao, J.; Song, J.; Nikolic-Paterson, D.J.; Li, J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 2012, 228, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.D.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiram, R.; Xiong, F.; Naud, P.; Xiao, J.; Sirois, M.; Tanguay, J.-F.; Tardif, J.-C.; Nattel, S. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc. Res. 2020, 117, 1776–1789. [Google Scholar] [CrossRef]
- Hiram, R. Resolution-promoting autacoids demonstrate promising cardioprotective effects against heart diseases. Mol. Biol. Rep. 2022, 1–19, ahead of print. [Google Scholar] [CrossRef]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef]
- Gesualdo, M.; Scicchitano, P.; Carbonara, S.; Ricci, G.; Principi, M.; Ierardi, E.; Di Leo, A.; Cortese, F.; Ciccone, M.M. The association between cardiac and gastrointestinal disorders: Causal or casual link? J. Cardiovasc. Med. 2016, 17, 330–338. [Google Scholar] [CrossRef]
- Potpara, T.S.; Jokic, V.; Dagres, N.; Marin, F.; Prostran, M.S.; Blomstrom-Lundqvist, C.; Lip, G.Y. Cardiac Arrhythmias in Patients with Chronic Kidney Disease: Implications of Renal Failure for Antiarrhythmic Drug Therapy. Curr. Med. Chem. 2016, 23, 2070–2083. [Google Scholar] [CrossRef]
- Mozos, I. Arrhythmia risk in liver cirrhosis. World J. Hepatol. 2015, 7, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Arenas de Larriva, A.P. Atrial Fibrillation, Cognitive Decline and Dementia. Eur. Cardiol. 2016, 11, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Lazzerini, P.E.; Laghi-Pasini, F.; Boutjdir, M.; Capecchi, P.L. Inflammatory cytokines and cardiac arrhythmias: The lesson from COVID-19. Nat. Rev. Immunol. 2022, 1–3, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. Systemic inflammation and arrhythmic risk: Lessons from rheumatoid arthritis. Eur. Heart J. 2016, 38, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Dobrev, D.; Nattel, S. Postoperative Atrial Fibrillation: Features, Mechanisms, and Clinical Management. Card. Electrophysiol. Clin. 2021, 13, 123–132. [Google Scholar] [CrossRef]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef]
- Carballo, S.; Pfenniger, A.; Carballo, D.; Garin, N.; James, R.W.; Mach, F.; Shah, D.; Kwak, B.R. Differential Association of Cx37 and Cx40 Genetic Variants in Atrial Fibrillation with and without Underlying Structural Heart Disease. Int. J. Mol. Sci. 2018, 19, 295. [Google Scholar] [CrossRef] [Green Version]
- Nofi, C.; Zhang, K.; Tang, Y.D.; Li, Y.; Migirov, A.; Ojamaa, K.; Gerdes, A.M.; Zhang, Y. Chronic dantrolene treatment attenuates cardiac dysfunction and reduces atrial fibrillation inducibility in a rat myocardial infarction heart failure model. Heart Rhythm O2 2020, 1, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Santulli, G.; Guerra, G.; Trotta, M.C.; Santamaria, M.; Sacra, C.; Testa, N.; Ducceschi, V.; Gatta, G.; Amico, M.D.; et al. Modulation of SERCA in Patients with Persistent Atrial Fibrillation Treated by Epicardial Thoracoscopic Ablation: The CAMAF Study. J. Clin. Med. 2020, 9, 544. [Google Scholar] [CrossRef] [Green Version]
- Nso, N.; Bookani, K.R.; Metzl, M.; Radparvar, F. Role of inflammation in atrial fibrillation: A comprehensive review of current knowledge. J. Arrhythmia 2020, 37, 1–10. [Google Scholar] [CrossRef]
- Silva, A.C.; Pereira, C.; Fonseca, A.C.R.G.; Pinto-Do-Ó, P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2021, 8, 1705. [Google Scholar] [CrossRef]
- Deb, A.; Ubil, E. Cardiac fibroblast in development and wound healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.M.; Villar, K.N.B.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell. Signal. 2020, 77, 109824. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1863, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Brás, L.E.D.C.; Toba, H.; Iyer, R.P.; Hall, M.E.; Winniford, M.D.; Lange, R.A.; Tyagi, S.C.; Lindsey, M.L. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-U’Datt, D.G.; Allen, B.G.; Hiram, R.; Alrabadi, N. Current knowledge into the role of the peptidylarginine deiminase (PAD) enzyme family in cardiovascular disease. Eur. J. Pharmacol. 2020, 891, 173765. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tang, X.; Liu, B.; Zhang, J.; Zhang, Y.; Lv, H.; Liu, D.; Mehta, J.L.; Wang, X. LOX-1 Deletion Attenuates Myocardial Fibrosis in the Aged Mice, Particularly Those with Hypertension. Front. Cardiovasc. Med. 2021, 8, 1305. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin. Med. Insights Cardiol. 2016, 10, CMC.S33164. [Google Scholar] [CrossRef] [Green Version]
- Richardson, W.J.; Clarke, S.A.; Quinn, T.A.; Holmes, J.W. Physiological Implications of Myocardial Scar Structure. Compr. Physiol. 2015, 5, 1877–1909. [Google Scholar] [CrossRef] [Green Version]
- Silvis, M.J.M.; Dengler, S.E.K.G.; Odille, C.A.; Mishra, M.; Van Der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; De Kleijn, D.P.V.; De Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 3135. [Google Scholar] [CrossRef]
- Rivera-Serrano, E.E.; Sherry, B. NF-κB activation is cell type-specific in the heart. Virology 2017, 502, 133–143. [Google Scholar] [CrossRef]
- Carrillo-Salinas, F.J.; Ngwenyama, N.; Anastasiou, M.; Kaur, K.; Alcaide, P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am. J. Pathol. 2019, 189, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Butcher, J.T. Inflammatory Regulation of Valvular Remodeling: The Good(?), the Bad, and the Ugly. Int. J. Inflamm. 2011, 2011, 721419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The Importance of NLRP3 Inflammasome in Heart Failure. J. Card. Fail. 2015, 21, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [Green Version]
- Toldo, S.; Kannan, H.; Bussani, R.; Anzini, M.; Sonnino, C.; Sinagra, G.; Merlo, M.; Mezzaroma, E.; De Giorgio, F.; Silvestri, F.; et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014, 171, e119–e121. [Google Scholar] [CrossRef]
- Louwe, M.C.; Olsen, M.B.; Kaasbøll, O.J.; Yang, K.; Fosshaug, L.E.; Alfsnes, K.; Øgaard, J.D.; Rashidi, A.; Skulberg, V.M.; Yang, M.; et al. Absence of NLRP3 Inflammasome in Hematopoietic Cells Reduces Adverse Remodeling after Experimental Myocardial Infarction. JACC Basic Transl. Sci. 2020, 5, 1210–1224. [Google Scholar] [CrossRef]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2019, 37, e174–e177. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Nakahira, K.; Dalli, J.; Siempos, I.I.; Norris, P.C.; Colas, R.A.; Moon, J.-S.; Shinohara, M.; Hisata, S.; Howrylak, J.A.; et al. NLRP3 Inflammasome Deficiency Protects against Microbial Sepsis via Increased Lipoxin B4 Synthesis. Am. J. Respir. Crit. Care Med. 2017, 196, 713–726. [Google Scholar] [CrossRef]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) Pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Szekely, Y.; Arbel, Y. A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today? Cardiol. Ther. 2018, 7, 25–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N. Interleukin-1 Receptor Type I Signaling Critically Regulates Infarct Healing and Cardiac Remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Zhao, L.; Pan, X.; Chen, N.N.; Chen, J.; Gong, Q.L.; Su, F.; Yan, J.; Zhang, Y.; Zhang, S.H. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-β1 signaling pathway. Lab. Investig. 2016, 96, 839–852, Erratum in Lab. Investig. 2016, 96, 1035. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.; Srivastava, A.; Watson, L.; Quinlan, L.R.; Pandit, A. Hyaluronic acid decreases IL-6 and IL-8 secretion and permeability in an inflammatory model of interstitial cystitis. Acta Biomater. 2015, 19, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Vistejnova, L.; Safrankova, B.; Nesporova, K.; Slavkovsky, R.; Hermannova, M.; Hosek, P.; Velebny, V.; Kubala, L. Low molecular weight hyaluronan mediated CD44 dependent induction of IL-6 and chemokines in human dermal fibroblasts potentiates innate immune response. Cytokine 2014, 70, 97–103. [Google Scholar] [CrossRef]
- Müller, J.; Gorressen, S.; Grandoch, M.; Feldmann, K.; Kretschmer, I.; Lehr, S.; Ding, Z.; Schmitt, J.P.; Schrader, J.; Garbers, C.; et al. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res. Cardiol. 2014, 109, 440. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer, D.S.; Patel, S. Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients with an Acute Coronary Syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Ni, H.; Huang, J.; Yu, G.; Zhang, Z.; Zhang, Q. VX-765 prevents intestinal ischemia-reperfusion injury by inhibiting NLRP3 inflammasome. Tissue Cell 2021, 75, 101718. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sharkawy, L.Y.; Brough, D.; Freeman, S. Inhibiting the NLRP3 Inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Liu, S.; Pan, P.; Su, X.; Tan, H.; Wu, D.; Zhang, L.; Song, C.; Dai, M.; et al. Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol. Immunol. 2018, 99, 134–144. [Google Scholar] [CrossRef]
- Martin, J.; Kelly, D.J.; Mifsud, S.A.; Zhang, Y.; Cox, A.J.; See, F.; Krum, H.; Wilkinson-Berka, J.; Gilbert, R.E. Tranilast attenuates cardiac matrix deposition in experimental diabetes: Role of transforming growth factor-beta. Cardiovasc. Res. 2005, 65, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Capulzini, L.; Brugada, P.; Brugada, J.; Brugada, R. Arrhythmia and Right Heart Disease: From Genetic Basis to Clinical Practice. Rev. Esp. Cardiol. 2010, 63, 963–983. [Google Scholar] [CrossRef]
- Churchill, T.W.; Baggish, A.L. The Right Heart: Acute and Chronic Issues. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 83. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Goedemans, L.; Leung, M.; van der Bijl, P.; Abou, R.; Vo, N.M.; Marsan, N.A.; Delgado, V.; Bax, J.J. Influence of Chronic Obstructive Pulmonary Disease on Atrial Mechanics by Speckle Tracking Echocardiography in Patients with Atrial Fibrillation. Am. J. Cardiol. 2020, 143, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Hiram, R.; Provencher, S. Pulmonary Disease, Pulmonary Hypertension and Atrial Fibrillation. Card. Electrophysiol. Clin. 2021, 13, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.-F.; Dohmen, P.M.; Choi, Y.-H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiram, R.; Naud, P.; Xiong, F.; Al-U’Datt, D.; Algalarrondo, V.; Sirois, M.G.; Tanguay, J.-F.; Tardif, J.-C.; Nattel, S. Right Atrial Mechanisms of Atrial Fibrillation in a Rat Model of Right Heart Disease. J. Am. Coll. Cardiol. 2019, 74, 1332–1347. [Google Scholar] [CrossRef] [PubMed]
- Medi, C.; Kalman, J.M.; Ling, L.-H.; Teh, A.W.; Lee, G.; Lee, G.; Spence, S.J.; Kaye, D.M.; Kistler, P.M. Atrial Electrical and Structural Remodeling Associated with Longstanding Pulmonary Hypertension and Right Ventricular Hypertrophy in Humans. J. Cardiovasc. Electrophysiol. 2012, 23, 614–620. [Google Scholar] [CrossRef]
- Schüttler, D.; Bapat, A.; Kääb, S.; Lee, K.; Tomsits, P.; Clauss, S.; Hucker, W.J. Animal Models of Atrial Fibrillation. Circ. Res. 2020, 127, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, A.; Garan, H.; Biviano, A. Arrhythmias in Pulmonary Arterial Hypertension. Prog. Cardiovasc. Dis. 2012, 55, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Benoist, D.; Stones, R.; Drinkhill, M.J.; Benson, A.P.; Yang, Z.; Cassan, C.; Gilbert, S.H.; Saint, D.A.; Cazorla, O.; Steele, D.S.; et al. Cardiac arrhythmia mechanisms in rats with heart failure induced by pulmonary hypertension. Am. J. Physiol. Circ. Physiol. 2012, 302, H2381–H2395. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Vanderpool, R.R.; Henry, B.L.; Palma, J.B.; Gabris, B.; Lai, Y.-C.; Hu, J.; Tofovic, S.P.; Reddy, R.P.; Mora, A.L.; et al. Relaxin Inhibits Ventricular Arrhythmia and Asystole in Rats with Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2021, 8, 663. [Google Scholar] [CrossRef]
- Andersen, A.; van der Feen, D.E.; Andersen, S.; Schultz, J.G.; Hansmann, G.; Bogaard, H.J. Animal models of right heart failure. Cardiovasc. Diagn. Ther. 2020, 10, 1561–1579. [Google Scholar] [CrossRef]
- Hessel, M.H.M.; Steendijk, P.; Adel, B.D.; Schutte, C.I.; Van Der Laarse, A. Characterization of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact rat. Am. J. Physiol. Circ. Physiol. 2006, 291, H2424–H2430. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Gamble, W.; Nadas, A.S.; Miettinen, O.S.; Reid, L. Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am. J. Physiol. Circ. Physiol. 1979, 236, H818–H827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taraseviciene-Stewart, L.; Nicolls, M.R.; Kraskauskas, D.; Scerbavicius, R.; Burns, N.; Cool, C.; Wood, K.; Parr, J.E.; Boackle, S.A.; Voelkel, N.F. Absence of T Cells Confers Increased Pulmonary Arterial Hypertension and Vascular Remodeling. Am. J. Respir. Crit. Care Med. 2007, 175, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Varian, K.D.; Bal, N.C.; Abraham, J.L.; Periasamy, M.; Janssen, P.M.L. Pulmonary artery banding alters the expression of Ca2+ transport proteins in the right atrium in rabbits. Am. J. Physiol. Circ. Physiol. 2009, 296, H1933–H1939. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, Y.; Okumura, K.; Ishii, R.; Slorach, C.; Hui, W.; Ide, H.; Honjo, O.; Sun, M.; Kabir, M.G.; Connelly, K.; et al. Pulmonary artery banding is a relevant model to study the right ventricular remodeling and dysfunction that occurs in pulmonary arterial hypertension. J. Appl. Physiol. 2020, 129, 238–246. [Google Scholar] [CrossRef]
- Arias-Loza, P.-A.; Jung, P.; Abeßer, M.; Umbenhauer, S.; Williams, T.; Frantz, S.; Schuh, K.; Pelzer, T. Development and Characterization of an Inducible Rat Model of Chronic Thromboembolic Pulmonary Hypertension. Hypertension 2016, 67, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Kong, B.; Dai, C.; Xiao, Z.; Fang, J.; Shuai, W.; Huang, H. Protective effects of Dapagliflozin on the vulnerability of ventricular arrhythmia in rats with pulmonary artery hypertension induced by monocrotaline. Bioengineered 2022, 13, 2697–2709. [Google Scholar] [CrossRef]
- Vitali, S.H.; Hansmann, G.; Rose, C.; Fernandez-Gonzalez, A.; Scheid, A.; Mitsialis, S.A.; Kourembanas, S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: Long-term follow-up. Pulm. Circ. 2014, 4, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.L.; Gabris, B.; Li, Q.; Martin, B.; Giannini, M.; Parikh, A.; Patel, D.; Haney, J.; Schwartzman, D.S.; Shroff, S.G.; et al. Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na+ channels. Heart Rhythm. 2016, 13, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.; Patel, D.; McTiernan, C.F.; Xiang, W.; Haney, J.; Yang, L.; Lin, B.; Kaplan, A.D.; Bett, G.; Rasmusson, R.L.; et al. Relaxin Suppresses Atrial Fibrillation by Reversing Fibrosis and Myocyte Hypertrophy and Increasing Conduction Velocity and Sodium Current in Spontaneously Hypertensive Rat Hearts. Circ. Res. 2013, 113, 313–321. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medication | Role on IL1β or/and NLRP3 Inflammasome | Ongoing Clinical Trials | ||
|---|---|---|---|---|
| Mode of Action | References | Clinical Trial Theme | Clinical Trial ID | |
| Belnacasan | CASP-1 specific inhibitor, Prevents Il-1 β release | [102] | SARS-CoV-2 | NCT05164120 |
| Canakinumab | Inhibits IL-1β | [101] | Inflammation and Cardiovascular risk | NCT02272946 |
| Colchicine | Decreases IL-1β, IL-18, and IL-6 | [100] | Peripheral artery disease | NCT04774159 |
| Perioperative AF | NCT03310125 | |||
| Dapansutrile | Blocks NLRP3 assembly, Prevents NLRP3-induced release of IL-1β and IL-18 | [103] | Schnitzler’s syndrome | NCT03595371 |
| NCT04540120 | ||||
| Inzomelid | Prevents ASC release and NLRP3 activation | [104] | Cryopryrin-Associated Periodic syndromes | EudraCT2020-000489-40 |
| Parkinson’s disease | NCT04338997 | |||
| Pirfenidone | Decreases NLRP3 and ASC expression Inhibits caspase-1 activation and Prevents IL-1β maturation | [105] | Pulmonary Fibrosis | NCT03109288 |
| NCT03109288 | ||||
| SGLT2 inhibitors | Prevents IL-1β release | [107] | Myocardial Infarction | NCT04899479 |
| Somalix | Inhibits NLRP3 activation | [104] | Peripheral arterial disease | NCT04015076 |
| Tranilast | Binds NACHT domain of NLRP3 to block NLRP3 oligonerization | [106] | Mucinoses | NCT03490708 |
| Cryopryrin-Associated Periodic syndromes | NCT03923140 | |||
| Experimental Models of RHD | Resolution Strategies | Anti-Arrhythmogenic Effects | References |
|---|---|---|---|
| Monocrotaline (MCT) | Resolvin D1 (RvD1) (i.p.: 2 µg/kg/d; 3w) | • ↓ Atrial fibrosis • ↓ Expression of IL6, TGF-β, ICAM1, IL1β NLRP3-inflammasome • ↑ Expression of IL10, CHEMR23 • ↓ AF susceptibility | Hiram et al., 2021 [54] |
| Dapagliflozin (DA) (per os: 60 mg/L; 4w) | • ↓ Ventricular fibrosis • Prevented channelopathy • ↓ TLR4 and NFκB activity • ↓ VF vulnerability | Qin et al., 2022 [127] | |
| Sugen-Hypoxia (SuHx) | Relaxin (RLX) (sc.: 30–400 µg/kg/d; 6w) | • ↓ NRF2 and gluthathione transferase • ↓ Expression of TGF-β, MMP2, MMP9, COLI and COLIII • ↓ Ventricular fibrosis and VF • ↓ Atrial fibrosis and AF | Martin et al., 2021 [119] Parikh et al., 2013 [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Younes, R.; LeBlanc, C.-A.; Hiram, R. Evidence of Failed Resolution Mechanisms in Arrhythmogenic Inflammation, Fibrosis and Right Heart Disease. Biomolecules 2022, 12, 720. https://doi.org/10.3390/biom12050720
Younes R, LeBlanc C-A, Hiram R. Evidence of Failed Resolution Mechanisms in Arrhythmogenic Inflammation, Fibrosis and Right Heart Disease. Biomolecules. 2022; 12(5):720. https://doi.org/10.3390/biom12050720
Chicago/Turabian StyleYounes, Rim, Charles-Alexandre LeBlanc, and Roddy Hiram. 2022. "Evidence of Failed Resolution Mechanisms in Arrhythmogenic Inflammation, Fibrosis and Right Heart Disease" Biomolecules 12, no. 5: 720. https://doi.org/10.3390/biom12050720
APA StyleYounes, R., LeBlanc, C. -A., & Hiram, R. (2022). Evidence of Failed Resolution Mechanisms in Arrhythmogenic Inflammation, Fibrosis and Right Heart Disease. Biomolecules, 12(5), 720. https://doi.org/10.3390/biom12050720