
Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction


Abstract
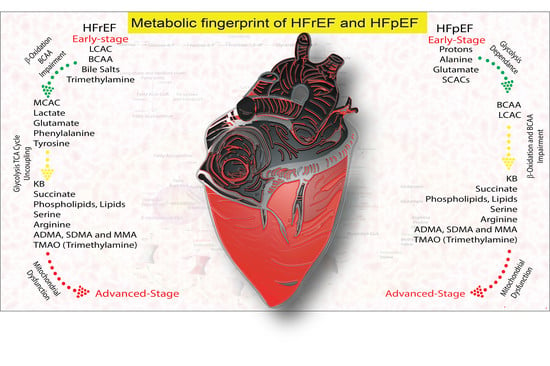
:
1. Introduction
1.1. Cardiac Metabolism of HFpEF and HFrEF and Preclinical Models
1.2. Fatty Acids Uptake and Oxidation
1.3. Ketone Bodies and Short-Chain Acyl Carnitines
1.4. Branched-Chain Amino Acids Oxidation
1.5. Glycolysis and Glucose Oxidation
1.6. Mitochondrial Dysfunction
1.7. The Microbiota Effect
1.8. Metabolomic Fingerprinting and Clinical Perspective
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Suthahar, N.; Meijers, W.C.; Ho, J.E.; Gansevoort, R.T.; Voors, A.A.; van der Meer, P.; Bakker, S.J.L.; Heymans, S.; van Empel, V.; Schroen, B.; et al. Sex-specific associations of obesity and N-terminal pro-B-type natriuretic peptide levels in the general population. Eur. J. Heart Fail 2018, 20, 1205–1214. [Google Scholar]
- Henkens, M.; Remmelzwaal, S.; Robinson, E.L.; van Ballegooijen, A.J.; Barandiarán Aizpurua, A.; Verdonschot, J.A.J.; Raafs, A.G.; Weerts, J.; Hazebroek, M.R.; Sanders-van Wijk, S.; et al. Risk of bias in studies investigating novel diagnostic biomarkers for heart failure with preserved ejection fraction. A systematic review. Eur. J. Heart Fail 2020, 22, 1586–1597. [Google Scholar]
- Zile, M.R.; Baicu, C.F. Biomarkers of diastolic dysfunction and myocardial fibrosis: Application to heart failure with a preserved ejection fraction. J. Cardiovasc. Transl. Res. 2013, 6, 501–515. [Google Scholar]
- Noordali, H.; Loudon, B.L.; Frenneaux, M.P.; Madhani, M. Cardiac metabolism-A promising therapeutic target for heart failure. Pharmacol. Ther. 2018, 182, 95–114. [Google Scholar]
- White, P.J.; McGarrah, R.W.; Grimsrud, P.A.; Tso, S.C.; Yang, W.H.; Haldeman, J.M.; Grenier-Larouche, T.; An, J.; Lapworth, A.L.; Astapova, I.; et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 2018, 27, 1281–1293. [Google Scholar]
- Hu, H.; Jaskiewicz, J.A.; Harris, R.A. Ethanol and oleate inhibition of α-ketoisovalerate and 3-hydroxyisobutyrate metabolism by isolated hepatocytes. Arch. Biochem. Biophys. 1992, 299, 57–62. [Google Scholar]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and molecular differences between HFpEF and HFrEF: A step ahead in an improved pathological understanding. Cells 2020, 9, 242. [Google Scholar]
- Contessotto, P.; Orbanić, D.; Da Costa, M.; Jin, C.; Owens, P.; Chantepie, S.; Chinello, C.; Newell, J.; Magni, F.; Papy-Garcia, D.; et al. Elastin-like recombinamers-based hydrogel modulates post-ischemic remodeling in a non-transmural myocardial infarction in sheep. Sci. Transl. Med. 2021, 13, eaaz5380. [Google Scholar]
- Contessotto, P.; Pandit, A. Therapies to prevent post-infarction remodelling: From repair to regeneration. Biomaterials 2021, 275, 120906. [Google Scholar]
- Li, H.; Xia, Y.Y.; Xia, C.L.; Li, Z.; Shi, Y.; Li, X.B.; Zhang, J.X. Mimicking metabolic disturbance in establishing animal models of heart failure with preserved ejection fraction. Front. Physiol. 2022, 13, 879214. [Google Scholar]
- Yoon, S.; Eom, G.H. Heart failure with preserved ejection fraction: Present status and future directions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar]
- Conceição, G.; Heinonen, I.; Lourenço, A.P.; Duncker, D.J.; Falcão-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 275–286. [Google Scholar]
- Degens, H.; de Brouwer, K.F.; Gilde, A.J.; Lindhout, M.; Willemsen, P.H.; Janssen, B.J.; van der Vusse, G.J.; van Bilsen, M. Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res. Cardiol. 2006, 101, 17–26. [Google Scholar]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar]
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Van Tassell, B.; Natarajan, R.; dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ. Heart Fail 2013, 6, 136–144. [Google Scholar]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail 2014, 7, 1022–1031. [Google Scholar]
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit, G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1103–H1113. [Google Scholar]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail 2012, 5, 493–503. [Google Scholar]
- Mori, J.; Zhang, L.; Oudit, G.Y.; Lopaschuk, G.D. Impact of the renin-angiotensin system on cardiac energy metabolism in heart failure. J. Mol. Cell Cardiol. 2013, 63, 98–106. [Google Scholar]
- Pellieux, C.; Aasum, E.; Larsen, T.S.; Montessuit, C.; Papageorgiou, I.; Pedrazzini, T.; Lerch, R. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J. Mol. Cell Cardiol. 2006, 41, 459–466. [Google Scholar]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar]
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035. [Google Scholar]
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Nagahata-Naito, Y.; Obinata, H.; Sano, M.; Sunaga, H.; et al. Glucose is preferentially utilized for biomass synthesis in pressure-overloaded hearts: Evidence from fatty acid-binding protein-4 and -5 knockout mice. Cardiovasc. Res. 2018, 114, 1132–1144. [Google Scholar]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: Evidence for shared metabolic impairments in clinical heart failure. J. Am. Heart Assoc. 2016, 5, e003190. [Google Scholar]
- Zhao, H.; Shui, B.; Zhao, Q.; Hu, Z.; Shu, Q.; Su, M.; Zhang, Y.; Ni, Y. Quantitative metabolomics reveals heart failure with midrange ejection fraction as a distinct phenotype of heart failure. Can. J. Cardiol. 2021, 37, 300–309. [Google Scholar]
- Cheng, M.L.; Wang, C.H.; Shiao, M.S.; Liu, M.H.; Huang, Y.Y.; Huang, C.Y.; Mao, C.T.; Lin, J.F.; Ho, H.Y.; Yang, N.I. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: Diagnostic and prognostic value of metabolomics. J. Am. Coll. Cardiol. 2015, 65, 1509–1520. [Google Scholar]
- Chen, W.S.; Liu, M.H.; Cheng, M.L.; Wang, C.H. Decreases in circulating concentrations of short-chain acylcarnitines are associated with systolic function improvement after decompensated heart failure. Int. Heart J. 2020, 61, 1014–1021. [Google Scholar]
- Hage, C.; Löfgren, L.; Michopoulos, F.; Nilsson, R.; Davidsson, P.; Kumar, C.; Ekström, M.; Eriksson, M.J.; Lyngå, P.; Persson, B.; et al. Metabolomic profile in HFpEF vs HFrEF patients. J. Card. Fail 2020, 26, 1050–1059. [Google Scholar]
- Zordoky, B.N.; Sung, M.M.; Ezekowitz, J.; Mandal, R.; Han, B.; Bjorndahl, T.C.; Bouatra, S.; Anderson, T.; Oudit, G.Y.; Wishart, D.S.; et al. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS ONE 2015, 10, e0124844. [Google Scholar]
- Wang, H.; Anstrom, K.; Ilkayeva, O.; Muehlbauer, M.J.; Bain, J.R.; McNulty, S.; Newgard, C.B.; Kraus, W.E.; Hernandez, A.; Felker, G.M.; et al. Sildenafil treatment in heart failure with preserved ejection fraction: Targeted metabolomic profiling in the relax trial. JAMA Cardiol. 2017, 2, 896–901. [Google Scholar]
- Tourki, B.; Kain, V.; Shaikh, S.R.; Leroy, X.; Serhan, C.N.; Halade, G.V. Deficit of resolution receptor magnifies inflammatory leukocyte directed cardiorenal and endothelial dysfunction with signs of cardiomyopathy of obesity. FASEB J. 2020, 34, 10560–10573. [Google Scholar]
- Ferro, F.; Ouillé, A.; Tran, T.A.; Fontanaud, P.; Bois, P.; Babuty, D.; Labarthe, F.; Le Guennec, J.Y. Long-chain acylcarnitines regulate the hERG channel. PLoS ONE 2012, 7, e41686. [Google Scholar]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P.; et al. Prognostic implications of long-chain acylcarnitines in heart failure and reversibility with mechanical circulatory support. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar]
- Aitken-Buck, H.M.; Krause, J.; Zeller, T.; Jones, P.P.; Lamberts, R.R. Long-chain acylcarnitines and cardiac excitation-contraction coupling: Links to arrhythmias. Front. Physiol. 2020, 11, 577856. [Google Scholar]
- Albert, C.L.; Tang, W.H.W. Metabolic biomarkers in heart failure. Heart Fail Clin. 2018, 14, 109–118. [Google Scholar]
- Stride, N.; Larsen, S.; Hey-Mogensen, M.; Sander, K.; Lund, J.T.; Gustafsson, F.; Køber, L.; Dela, F. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur. J. Heart Fail 2013, 15, 150–157. [Google Scholar]
- Chen, X.F.; Chen, X.; Tang, X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin. Sci. 2020, 134, 657–676. [Google Scholar]
- Carley, A.N.; Maurya, S.K.; Fasano, M.; Wang, Y.; Selzman, C.H.; Drakos, S.G.; Lewandowski, E.D. Short-chain fatty acids outpace ketone oxidation in the failing heart. Circulation 2021, 143, 1797–1808. [Google Scholar]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The failing heart relies on ketone bodies as a fuel. Circulation 2016, 133, 698–705. [Google Scholar]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079. [Google Scholar]
- Karwi, Q.G.; Zhang, L.; Wagg, C.S.; Wang, W.; Ghandi, M.; Thai, D.; Yan, H.; Ussher, J.R.; Oudit, G.Y.; Lopaschuk, G.D. Targeting the glucagon receptor improves cardiac function and enhances insulin sensitivity following a myocardial infarction. Cardiovasc. Diabetol. 2019, 18, 1. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar]
- Schugar, R.C.; Moll, A.R.; André d’Avignon, D.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar]
- De Jong, K.A.; Lopaschuk, G.D. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 2016, 133, 706–716. [Google Scholar]
- Du, Z.; Shen, A.; Huang, Y.; Su, L.; Lai, W.; Wang, P.; Xie, Z.; Xie, Z.; Zeng, Q.; Ren, H.; et al. 1H-NMR-based metabolic analysis of human serum reveals novel markers of myocardial energy expenditure in heart failure patients. PLoS ONE 2014, 9, e88102. [Google Scholar]
- Flores-Guerrero, J.L.; Westenbrink, B.D.; Connelly, M.A.; Otvos, J.D.; Groothof, D.; Shalaurova, I.; Garcia, E.; Navis, G.; de Boer, R.A.; Bakker, S.J.L.; et al. Association of beta-hydroxybutyrate with development of heart failure: Sex differences in a Dutch population cohort. Eur J. Clin. Investig. 2021, 51, e13468. [Google Scholar]
- Lommi, J.; Kupari, M.; Koskinen, P.; Näveri, H.; Leinonen, H.; Pulkki, K.; Härkönen, M. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol. 1996, 28, 665–672. [Google Scholar]
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased cardiac uptake of ketone bodies and free fatty acids in human heart failure and hypertrophic left ventricular remodeling. Circ. Heart Fail 2018, 11, e004953. [Google Scholar]
- Yokokawa, T.; Yoshihisa, A.; Kanno, Y.; Abe, S.; Misaka, T.; Yamada, S.; Kaneshiro, T.; Sato, T.; Oikawa, M.; Kobayashi, A.; et al. Circulating acetoacetate is associated with poor prognosis in heart failure patients. Int. J. Cardiol. Heart Vasc. 2019, 25, 100432. [Google Scholar]
- Andersson, C.; Liu, C.; Cheng, S.; Wang, T.J.; Gerszten, R.E.; Larson, M.G.; Vasan, R.S. Metabolomic signatures of cardiac remodelling and heart failure risk in the community. ESC Heart Fail 2020, 7, 3707–3715. [Google Scholar]
- Lommi, J.; Koskinen, P.; Näveri, H.; Härkönen, M.; Kupari, M. Heart failure ketosis. J. Intern. Med. 1997, 242, 231–238. [Google Scholar]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar]
- Stryeck, S.; Gastrager, M.; Degoricija, V.; Trbušić, M.; Potočnjak, I.; Radulović, B.; Pregartner, G.; Berghold, A.; Madl, T.; Frank, S. Serum concentrations of citrate, tyrosine, 2- and 3- hydroxybutyrate are associated with increased 3-month mortality in acute heart failure patients. Sci Rep. 2019, 9, 6743. [Google Scholar]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar]
- Fillmore, N.; Wagg, C.S.; Zhang, L.; Fukushima, A.; Lopaschuk, G.D. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1046–E1052. [Google Scholar]
- Li, R.; He, H.; Fang, S.; Hua, Y.; Yang, X.; Yuan, Y.; Liang, S.; Liu, P.; Tian, Y.; Xu, F.; et al. Time series characteristics of serum branched-chain amino acids for early diagnosis of chronic heart failure. J. Proteome Res. 2019, 18, 2121–2128. [Google Scholar]
- Sansbury, B.E.; DeMartino, A.M.; Xie, Z.; Brooks, A.C.; Brainard, R.E.; Watson, L.J.; DeFilippis, A.P.; Cummins, T.D.; Harbeson, M.A.; Brittian, K.R.; et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ. Heart Fail 2014, 7, 634–642. [Google Scholar]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 2016, 133, 2038–2049. [Google Scholar]
- Guo, N.; Yang, D.; Wang, X.; Dai, J.; Wang, M.; Lei, Y. Metabonomic study of chronic heart failure and effects of chinese herbal decoction in rats. J. Chromatogr. A 2014, 1362, 89–101. [Google Scholar]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar]
- Kadota, Y.; Toyoda, T.; Hayashi-Kato, M.; Kitaura, Y.; Shimomura, Y. Octanoic acid promotes branched-chain amino acid catabolisms via the inhibition of hepatic branched-chain alpha-keto acid dehydrogenase kinase in rats. Metabolism 2015, 64, 1157–1164. [Google Scholar]
- Saleem, T.H.; Algowhary, M.; Kamel, F.E.M.; El-Mahdy, R.I. Plasma amino acid metabolomic pattern in heart failure patients with either preserved or reduced ejection fraction: The relation to established risk variables and prognosis. Biomed. Chromatogr. 2020, 35, e5012. [Google Scholar]
- Wang, C.H.; Cheng, M.L.; Liu, M.H. Amino acid-based metabolic panel provides robust prognostic value additive to b-natriuretic peptide and traditional risk factors in heart failure. Dis. Markers 2018, 2018, 3784589. [Google Scholar]
- Deidda, M.; Piras, C.; Cadeddu Dessalvi, C.; Congia, D.; Locci, E.; Ascedu, F.; De Candia, G.; Cadeddu, M.; Lai, G.; Pirisi, R.; et al. Blood metabolomic fingerprint is distinct in healthy coronary and in stenosing or microvascular ischemic heart disease. J. Transl. Med. 2017, 15, 112. [Google Scholar]
- Chandler, M.P.; Kerner, J.; Huang, H.; Vazquez, E.; Reszko, A.; Martini, W.Z.; Hoppel, C.L.; Imai, M.; Rastogi, S.; Sabbah, H.N.; et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1538–H1543. [Google Scholar]
- Turer, A.; Altamirano, F.; Schiattarella, G.G.; May, H.; Gillette, T.G.; Malloy, C.R.; Merritt, M.E. Remodeling of substrate consumption in the murine sTAC model of heart failure. J. Mol. Cell. Cardiol. 2019, 134, 144–153. [Google Scholar]
- Zeng, H.; Chen, J.X. Sirtuin 3, endothelial metabolic reprogramming, and heart failure with preserved ejection fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, C.; Zaugg, M.; Lopaschuk, G.D. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ. Heart Fail 2013, 6, 1039–1048. [Google Scholar]
- Fernandez-Caggiano, M.; Eaton, P. Heart failure-emerging roles for the mitochondrial pyruvate carrier. Cell Death Differ. 2021, 28, 1149–1158. [Google Scholar]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar]
- Morimoto, S.; Goto, T. Role of troponin I isoform switching in determining the pH sensitivity of Ca2+ regulation in developing rabbit cardiac muscle. Biochem. Biophys. Res. Commun. 2000, 267, 912–917. [Google Scholar]
- Contaifer, D., Jr.; Buckley, L.F.; Wohlford, G.; Kumar, N.G.; Morriss, J.M.; Ranasinghe, A.D.; Carbone, S.; Canada, J.M.; Trankle, C.; Abbate, A.; et al. Metabolic modulation predicts heart failure tests performance. PLoS ONE 2019, 14, e0218153. [Google Scholar]
- Desmoulin, F.; Galinier, M.; Trouillet, C.; Berry, M.; Delmas, C.; Turkieh, A.; Massabuau, P.; Taegtmeyer, H.; Smih, F.; Rouet, P. Metabonomics analysis of plasma reveals the lactate to cholesterol ratio as an independent prognostic factor of short-term mortality in acute heart failure. PLoS ONE 2013, 8, e60737. [Google Scholar]
- Lazzeri, C.; Valente, S.; Chiostri, M.; Picariello, C.; Gensini, G.F. Lactate in the acute phase of ST-elevation myocardial infarction treated with mechanical revascularization: A single-center experience. Am. J. Emerg. Med. 2012, 30, 92–96. [Google Scholar]
- Shibayama, J.; Yuzyuk, T.N.; Cox, J.; Makaju, A.; Miller, M.; Lichter, J.; Li, H.; Leavy, J.D.; Franklin, S.; Zaitsev, A.V. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: Integrated metabolomics and proteomics study. PLoS ONE 2015, 10, e0118974. [Google Scholar]
- Sakao, S.; Kawakami, E.; Shoji, H.; Naito, A.; Miwa, H.; Suda, R.; Sanada, T.J.; Tanabe, N.; Tatsumi, K. Metabolic remodeling in the right ventricle of rats with severe pulmonary arterial hypertension. Mol. Med. Rep. 2021, 23, 1. [Google Scholar]
- Forsey, R.G.; Reid, K.; Brosnan, J.T. Competition between fatty acids and carbohydrate or ketone bodies as metabolic fuels for the isolated perfused heart. Can. J. Physiol. Pharmacol. 1987, 65, 401–406. [Google Scholar]
- Purmal, C.; Kucejova, B.; Sherry, A.D.; Burgess, S.C.; Malloy, C.R.; Merritt, M.E. Propionate stimulates pyruvate oxidation in the presence of acetate. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1134–H1141. [Google Scholar]
- Russell, R.R., 3rd; Taegtmeyer, H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J. Clin. Investig. 1991, 87, 384–390. [Google Scholar]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Kraus, W.E.; Shah, S.H. Metabolic dysfunction in heart failure: Diagnostic, prognostic, and pathophysiologic insights from metabolomic profiling. Curr. Heart Fail Rep. 2016, 13, 119–131. [Google Scholar]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar]
- Zhou, J.; Chen, X.; Chen, W.; Zhong, L.; Cui, M. Comprehensive plasma metabolomic and lipidomic analyses reveal potential biomarkers for heart failure. Mol. Cell. Biochem. 2021, 476, 3449–3460. [Google Scholar]
- Arsenian, M. Potential cardiovascular applications of glutamate, aspartate, and other amino acids. Clin. Cardiol. 1998, 21, 620–624. [Google Scholar]
- Zheng, Y.; Hu, F.B.; Ruiz-Canela, M.; Clish, C.B.; Dennis, C.; Salas-Salvado, J.; Hruby, A.; Liang, L.; Toledo, E.; Corella, D.; et al. Metabolites of glutamate metabolism are associated with incident cardiovascular events in the PREDIMED PREvención con Dieta MEDiterránea (PREDIMED) trial. J. Am. Heart Assoc. 2016, 5, e003755. [Google Scholar]
- Hiraiwa, H.; Okumura, T.; Kondo, T.; Kato, T.; Kazama, S.; Kimura, Y.; Ishihara, T.; Iwata, E.; Shimojo, M.; Kondo, S.; et al. Prognostic value of leucine/phenylalanine ratio as an amino acid profile of heart failure. Heart Vessel. 2021, 36, 965–977. [Google Scholar]
- Wang, C.H.; Cheng, M.L.; Liu, M.H. Simplified plasma essential amino acid-based profiling provides metabolic information and prognostic value additive to traditional risk factors in heart failure. Amino Acids 2018, 50, 1739–1748. [Google Scholar]
- Blain, A.M.; Straub, V.W. δ-Sarcoglycan-deficient muscular dystrophy: From discovery to therapeutic approaches. Skelet. Muscle 2011, 1, 13. [Google Scholar]
- Okumura, K.; Yamada, Y.; Kondo, J.; Hashimoto, H.; Ito, T.; Kitoh, J. Decreased 1,2-diacylglycerol levels in myopathic hamster hearts during the development of heart failure. J. Mol. Cell. Cardiol. 1991, 23, 409–416. [Google Scholar]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41. [Google Scholar]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic origins of heart failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar]
- Basu Ball, W.; Neff, J.K.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2018, 592, 1273–1290. [Google Scholar]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Acín-Pérez, R.; Cogliati, S.; López-Olañeta, M.; Ortiz-Sánchez, P.; Bonzón-Kulichenko, E.; Vázquez, J.; García-Pavía, P.; et al. Activation of serine one-carbon metabolism by calcineurin Aβ1 reduces myocardial hypertrophy and improves ventricular function. J. Am. Coll. Cardiol. 2018, 71, 654–667. [Google Scholar]
- Zhou, X.; He, L.; Zuo, S.; Zhang, Y.; Wan, D.; Long, C.; Huang, P.; Wu, X.; Wu, C.; Liu, G.; et al. Serine prevented high-fat diet-induced oxidative stress by activating AMPK and epigenetically modulating the expression of glutathione synthesis-related genes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 488–498. [Google Scholar]
- Shao, Z.; Wang, Z.; Shrestha, K.; Thakur, A.; Borowski, A.G.; Sweet, W.; Thomas, J.D.; Moravec, C.S.; Hazen, S.L.; Tang, W.H. Pulmonary hypertension associated with advanced systolic heart failure: Dysregulated arginine metabolism and importance of compensatory dimethylarginine dimethylaminohydrolase-1. J. Am. Coll. Cardiol. 2012, 59, 1150–1158. [Google Scholar]
- Tang, W.H.; Tong, W.; Shrestha, K.; Wang, Z.; Levison, B.S.; Delfraino, B.; Hu, B.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Differential effects of arginine methylation on diastolic dysfunction and disease progression in patients with chronic systolic heart failure. Eur. Heart J. 2008, 29, 2506–2513. [Google Scholar]
- O’Donovan, A.N.; Herisson, F.M.; Fouhy, F.; Ryan, P.M.; Whelan, D.; Johnson, C.N.; Cluzel, G.; Ross, R.P.; Stanton, C.; Caplice, N.M. Gut microbiome of a porcine model of metabolic syndrome and HF-pEF. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H590–H603. [Google Scholar]
- Guo, F.; Qiu, X.; Tan, Z.; Li, Z.; Ouyang, D. Plasma trimethylamine n-oxide is associated with renal function in patients with heart failure with preserved ejection fraction. BMC Cardiovasc. Disord. 2020, 20, 394. [Google Scholar]
- Schuett, K.; Kleber, M.E.; Scharnagl, H.; Lorkowski, S.; März, W.; Niessner, A.; Marx, N.; Meinitzer, A. Trimethylamine-N-oxide and heart failure with reduced versus preserved ejection fraction. J. Am. Coll. Cardiol. 2017, 70, 3202–3204. [Google Scholar]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How gut microbiota contributes to heart failure. Transl. Res. 2021, 228, 109–125. [Google Scholar]
- Naghipour, S.; Cox, A.J.; Peart, J.N.; Du Toit, E.F.; Headrick, J.P. Trimethylamine N-oxide: Heart of the microbiota-CVD nexus? Nutr. Res. Rev. 2021, 34, 125–146. [Google Scholar]
- Chhibber-Goel, J.; Gaur, A.; Singhal, V.; Parakh, N.; Bhargava, B.; Sharma, A. The complex metabolism of trimethylamine in humans: Endogenous and exogenous sources. Expert Rev. Mol. Med. 2016, 18, e8. [Google Scholar]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.M.; Gasecka, A.; Kaplon-Cieslicka, A.; Filipiak, K.; Sikora, E.; et al. TMA, A forgotten uremic toxin, but not TMAO, is involved in cardiovascular pathology. Toxins Basel 2019, 11, 490. [Google Scholar]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar]
- Pakhomov, N.; Baugh, J.A. The role of diet-derived short-chain fatty acids in regulating cardiac pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H475–H486. [Google Scholar]
- Beale, A.L.; O’Donnell, J.A.; Nakai, M.E.; Nanayakkara, S.; Vizi, D.; Carter, K.; Dean, E.; Ribeiro, R.V.; Yiallourou, S.; Carrington, M.J.; et al. The gut microbiome of heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2021, 10, e020654. [Google Scholar]
- Raufman, J.P.; Chen, Y.; Zimniak, P.; Cheng, K. Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology 2002, 65, 215–221. [Google Scholar]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar]
- Ljubuncic, P.; Said, O.; Ehrlich, Y.; Meddings, J.B.; Shaffer, E.A.; Bomzon, A. On the in vitro vasoactivity of bile acids. Br. J. Pharmacol. 2000, 131, 387–398. [Google Scholar]
- Mayerhofer, C.C.K.; Ueland, T.; Broch, K.; Vincent, R.P.; Cross, G.F.; Dahl, C.P.; Aukrust, P.; Gullestad, L.; Hov, J.R.; Trøseid, M. Increased secondary/primary bile acid ratio in chronic heart failure. J. Card. Fail. 2017, 23, 666–671. [Google Scholar]
- Eblimit, Z.; Thevananther, S.; Karpen, S.J.; Taegtmeyer, H.; Moore, D.D.; Adorini, L.; Penny, D.J.; Desai, M.S. TGR5 activation induces cytoprotective changes in the heart and improves myocardial adaptability to physiologic, inotropic, and pressure-induced stress in mice. Cardiovasc. Ther. 2018, 36, e12462. [Google Scholar]
- von Haehling, S.; Schefold, J.C.; Jankowska, E.A.; Springer, J.; Vazir, A.; Kalra, P.R.; Sandek, A.; Fauler, G.; Stojakovic, T.; Trauner, M.; et al. Ursodeoxycholic acid in patients with chronic heart failure: A double-blind, randomized, placebo-controlled, crossover trial. J. Am. Coll. Cardiol. 2012, 59, 585–592. [Google Scholar]
- Zhang, J.; Wang, L.; Cai, J.; Lei, A.; Liu, C.; Lin, R.; Jia, L.; Fu, Y. Gut microbial metabolite TMAO portends prognosis in acute ischemic stroke. J. Neuroimmunol. 2021, 354, 577526. [Google Scholar]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Protein turnover and amino acid catabolism. In Biochemistry, 8th ed.; W H Freeman: New York, NY, USA, 2007. [Google Scholar]
- Shao, D.; Villet, O.; Zhang, Z.; Choi, S.W.; Yan, J.; Ritterhoff, J.; Gu, H.; Djukovic, D.; Christodoulou, D.; Kolwicz, S.C., Jr.; et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat. Commun. 2018, 9, 2935. [Google Scholar]
- Wang, J.; Li, Z.; Chen, J.; Zhao, H.; Luo, L.; Chen, C.; Xu, X.; Zhang, W.; Gao, K.; Li, B.; et al. Metabolomic identification of diagnostic plasma biomarkers in humans with chronic heart failure. Mol. Biosyst. 2013, 9, 2618–2626. [Google Scholar]
- Tsuji, S.; Koyama, S.; Taniguchi, R.; Fujiwara, T.; Fujiwara, H.; Sato, Y. Nutritional status of outpatients with chronic stable heart failure based on serum amino acid concentration. J. Cardiol. 2018, 72, 458–465. [Google Scholar]
- Zhenyukh, O.; González-Amor, M.; Rodrigues-Diez, R.R.; Esteban, V.; Ruiz-Ortega, M.; Salaices, M.; Mas, S.; Briones, A.M.; Egido, J. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J. Cell. Mol. Med. 2018, 22, 4948–4962. [Google Scholar]
- Ussher, J.R.; Jaswal, J.S.; Lopaschuk, G.D. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ. Res. 2012, 111, 628–641. [Google Scholar]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar]
- Wang, C.H.; Cheng, M.L.; Liu, M.H.; Fu, T.C. Amino acid-based metabolic profile provides functional assessment and prognostic value for heart failure outpatients. Dis. Markers 2019, 2019, 8632726. [Google Scholar]
- Neubauer, S. The failing heart-an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar]
- Schwarz, K.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart-mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar]
- Stenemo, M.; Ganna, A.; Salihovic, S.; Nowak, C.; Sundström, J.; Giedraitis, V.; Broeckling, C.D.; Prenni, J.E.; Svensson, P.; Magnusson, P.K.E.; et al. The metabolites urobilin and sphingomyelin (30:1) are associated with incident heart failure in the general population. ESC Heart Fail. 2019, 6, 764–773. [Google Scholar]
- Lee, D.S.; Gona, P.; Vasan, R.S.; Larson, M.G.; Benjamin, E.J.; Wang, T.J.; Tu, J.V.; Levy, D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: Insights from the framingham heart study of the national heart, lung, and blood institute. Circulation 2009, 119, 3070–3077. [Google Scholar]
- Savji, N.; Meijers, W.C.; Bartz, T.M.; Bhambhani, V.; Cushman, M.; Nayor, M.; Kizer, J.R.; Sarma, A.; Blaha, M.J.; Gansevoort, R.T.; et al. The association of obesity and cardiometabolic traits with incident HFpEF and HFrEF. JACC Heart Fail. 2018, 6, 701–709. [Google Scholar]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting age-related pathways in heart failure. Circ. Res. 2020, 126, 533–551. [Google Scholar]
- Sandesara, P.B.; O’Neal, W.T.; Kelli, H.M.; Samman-Tahhan, A.; Hammadah, M.; Quyyumi, A.A.; Sperling, L.S. The prognostic significance of diabetes and microvascular complications in patients with heart failure with preserved ejection fraction. Diabetes Care 2018, 41, 150–155. [Google Scholar]
- Tomasoni, D.; Adamo, M.; Anker, M.S.; von Haehling, S.; Coats, A.J.S.; Metra, M. Heart failure in the last year: Progress and perspective. ESC Heart Fail. 2020, 7, 3505–3530. [Google Scholar]
- Sabbatini, A.R.; Kararigas, G. Menopause-related estrogen decrease and the pathogenesis of HFpEF: JACC review topic of the week. J. Am. Coll. Cardiol. 2020, 75, 1074–1082. [Google Scholar]
- Lam, C.S.P.; Arnott, C.; Beale, A.L.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.M.; Ky, B.; Santema, B.T.; Sliwa, K.; Voors, A.A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859–3868c. [Google Scholar]
- Ashrafian, H.; Sounderajah, V.; Glen, R.; Ebbels, T.; Blaise, B.J.; Kalra, D.; Kultima, K.; Spjuth, O.; Tenori, L.; Salek, R.M.; et al. Metabolomics: The stethoscope for the twenty-first century. Med. Princ. Pract. 2021, 30, 301–310. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite, Pathway | Early HFrEF | Adv HFrEF | Early HFpEF | Adv HFpEF | Physiological Effect | Pathological Effect | Ref. |
|---|---|---|---|---|---|---|---|
| Fatty Acids, Acylcarnitines oxidation | − − − | − − − | = | − − − | Fatty Acids β-Oxidation | FA accumulation | [15,16,17,18,19,20,21,22,23,24,25] |
| Long-Chain Acylcarnitines Oxidation | + | + | = | + | ATP production, ketone bodies formation, FA oxidation | Diabetes, reduced contractility, inflammation, arrhythmogenesis, lipotoxicity, ROS production, nitric oxide and ATP reduction | [26,27,28,29,31,32,33,34,35,36,37,38,39,40,41] |
| Medium-Chain Acylcarnitines Oxidation | + | + | = | + | ATP production, ketone bodies formation, FA oxidation | Transition to HF, lipotoxicity, ROS production, nitric oxide and ATP reduction | [28,30,32] |
| Short-Chain Acylcarnitines Oxidation | + | + | = | =/+ | ATP production, ketone bodies formation, FA metabolism | Diabetes, hypertension, ROS production, nitric oxide and ATP reduction | [26,30,31,42,43] |
| Ketone bodies oxidation | + | − − − | = | − − − | Ketone bodies oxidation | KBs accumulation | [43,44,45,46,47,48,49,50,51,52] |
| KBs Oxidation (Acetone, Acetoacetate, 3-Hydroxybutyrate) | + | − | = | − | ATP production, anti-inflammatory, epigenomic regulation | Hypertension, Inflammation, ROS production, nitric oxide and ATP reduction | [26,32,43,45,46,47,49,50,51,52,53,54,55,56,57,58,59,60,61] |
| Succinate | + | − | = | − | TCA cycle intermediate, ketone bodies formation, FA oxidation | Ischemia, inflammation, and hypoxic signaling, ROS production, nitric oxide and ATP reduction | [37,54,55,56,60,62,63,64] |
| Branched-Chain Amino Acids oxidation | − | − − − | = | − − | BCAA oxidation | BCAA accumulation | [5,18,65,66,67,68,69,70,71] |
| Leucine, Isoleucine, Valine | + | + | = | + | Anaplerotic reactions, ketone and short-chain fatty acids oxidation | Pro-anti-hypertrophic and pro-anti-inflammatory, FA accumulation | [5,18,28,29,32,54,71,72,73,74] |
| Glycolysis | = | + + + | + | + + + | Glucose anaerobic metabolism | Lactate and protons accumulation | [15,16,19,20,24,25,46,47,75,76,77,78,79,80,81] |
| Protons | = | + | + | + | ATP production | Reduced contractility, troponin I calcium binding, calcium current generation, and ATP availability | [15,16,77,81] |
| Lactate | + | + | = | =/− | Glycolysis, Glucose Oxidation | Myocardial infarction, contractile dysfunction, increased mortality | [54,59,63,82,83,84] |
| TCA cycle/anaplerotic reactions | − | − − − | = | − − − | Acetyl-CoA oxidation and TCA cycle intermediates replenishment | Reduced TCA cycle oxidative metabolism | [16,17,18,24,25,40,44,45,50,51,52,53,56,60,75,76,78,80,85,86,87,88,89] |
| Alanine | = | − | − | − | TCA cycle, anaplerotic reactions | Inflammation and ROS production | [31,54,72,74,90,91,92] |
| Glutamate | + | + | = | + | TCA cycle and anaplerotic reactions | Stroke, cardiovascular diseases | [29,72,73,93,94] |
| Phenylalanine | + | + | = | + | Glycolysis-glucose nitric oxide production, ketone bodies formation, anaplerotic reactions | Hypertension, reduced tissue perfusion, increased insulin resistance, increased protein breakdown, and hypoalbuminemia | [28,72,95] |
| Tyrosine | + | + | = | + | Glycolysis-glucose nitric oxide production, ketone bodies formation, anaplerotic reactions | Decreased synthesis of thyroid hormones, catecholamines, neurotransmitters, or serum proteins | [28,29,63,72,95,96] |
| Electron Transport Chain Oxidative Phosphorylation (ETC-OXPHOS) | = | − − − | − | − − − | ATP production | Reduced ATP production | [4,15,18,44,61,78,79,97,98,99,100,101] |
| Phosphatidylcholine, Lysophosphatidylcholine, Sphingomyelin | = | − | = | − | Membrane fluidity, contractility, cell signaling | Membrane stiffness, ROS production, nitric oxide and ATP reduction, apoptosis, inflammation, and ion channels dysregulation | [4,28,32,59,61,92,99,100,101,102] |
| Serine | = | − | = | − | Nitric oxide production | Oxidative stress, fibrosis | [31,72,73,103,104] |
| Arginine | = | − | = | − | Nitric oxide production, anaplerotic reactions | Reduced nitric oxide, hypertension | [28,31,105,106] |
| Dimethylarginine, Symmetric Dimethylarginine, and N-monomethylarginine | = | + | = | + | Nitric oxide production | Reduced nitric oxide, hypertension | [28,31,105,106] |
| Gut absorption and microbiota activity | +/− | +/− | +/− | +/− | Nutrients absorption | Production of metabolites with pathological effects | [42,43,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123] |
| Trimethylamine N-oxide, trimethylamine | = | + | = | + | Phosphatidylcholine, choline, and carnitine metabolism, chaperone, osmolyte, and piezolyte | Atherosclerosis and thrombosis, renal and liver function | [108,109,110,112,124] |
| Trimethylamine | + | + | = | + | TMAO precursor endogenous and esogenous | Obesity, diabetes, cardiovascular, and renal disorders | [113,114] |
| Short-chain fatty acids | = | + | =/− | + | ATP production, ketone bodies formation, FA metabolism | Hypertension, hypertrophy, and fibrosis | [42,43,110,115,116,117] |
| Bile acids | − | = | = | = | Vascular tone and blood pressure regulation, fat absorption, cholesterol, lipid, glucose metabolism | Hypertension | [110,118,119,120,121,122,123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferro, F.; Spelat, R.; Valente, C.; Contessotto, P. Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction. Biomolecules 2022, 12, 969. https://doi.org/10.3390/biom12070969
Ferro F, Spelat R, Valente C, Contessotto P. Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction. Biomolecules. 2022; 12(7):969. https://doi.org/10.3390/biom12070969
Chicago/Turabian StyleFerro, Federico, Renza Spelat, Camilla Valente, and Paolo Contessotto. 2022. "Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction" Biomolecules 12, no. 7: 969. https://doi.org/10.3390/biom12070969
APA StyleFerro, F., Spelat, R., Valente, C., & Contessotto, P. (2022). Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction. Biomolecules, 12(7), 969. https://doi.org/10.3390/biom12070969