
Adenine-Induced Nephropathy Reduces Atherosclerosis in ApoE Knockout Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice
2.3. Urine, Serum, and Tissue Biochemistry
2.4. Histology
2.5. RNA Extraction and Real-Time PCR in Kidney and Liver
2.6. MicroCT Scan
2.7. Cholesterol Efflux
2.8. Statistical Analysis
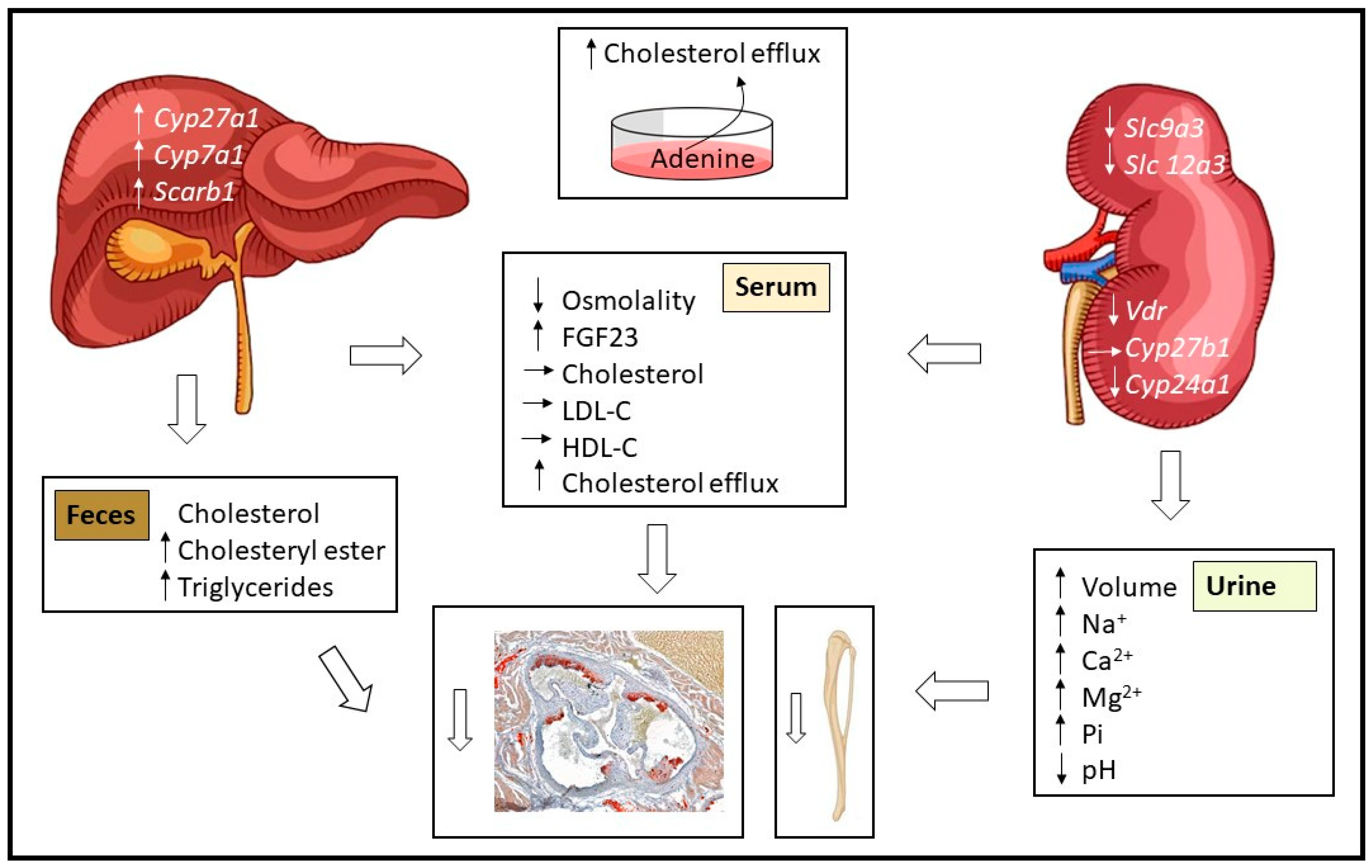
3. Results
3.1. Development of a New Model of Adenine-Induced Nephropathy with Mild Effect on Body Weight
3.2. The Anti-Atherosclerotic Effect of Adenine Is Mediated by Enhanced Cholesterol Efflux
3.3. Effect of Adenine on Hepatic Lipid Composition and Cholesterol Excretion
3.4. Impact of Adenine-Induced Nephropathy on Gene Expression in Kidney and Liver Tissues
3.5. Effect of Adenine on Bone Structure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goyal, A.; Yusuf, S. The burden of cardiovascular disease in the Indian subcontinent. Indian J. Med. Res. 2006, 124, 235–244. [Google Scholar] [PubMed]
- Barquera, S.; Pedroza-Tobías, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Fish, J.E. Transforming endothelial cells in atherosclerosis. Nat. Metab. 2019, 1, 856–857. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Psarros, C.; Demosthenous, M.; Patel, R.; Antoniades, C.; Stefanadis, C. Innate and adaptive inflammation as a therapeutic target in vascular disease: The emerging role of statins. J. Am. Coll. Cardiol. 2014, 63, 2491–2502. [Google Scholar] [CrossRef]
- Bruce, K.D.; Byrne, C.D. The metabolic syndrome: Common origins of a multifactorial disorder. Postgrad Med. J. 2009, 85, 614–621. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet and Lifestyle Drive the Pandemic of Obesity and Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [Google Scholar] [CrossRef]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Rohatgi, A. Reverse Cholesterol Transport and Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 2019, 39, 2–4. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Breslow, J.L. Mouse models of atherosclerosis. Science 1996, 272, 685–688. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Srivastava, N.; Averna, M. Dietary cholic acid lowers plasma levels of mouse and human apolipoprotein A-I primarily via a transcriptional mechanism. Eur. J. Biochem. 2000, 267, 4272–4280. [Google Scholar] [CrossRef]
- Murphy, C.; Parini, P.; Wang, J.; Björkhem, I.; Eggertsen, G.; Gåfvels, M. Cholic acid as key regulator of cholesterol synthesis, intestinal absorption and hepatic storage in mice. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2005, 1735, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kouri, A.M.; Rheault, M.N. Cardiovascular disease in children with chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2021, 30, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- McNerny, E.; Nickolas, T.L. Bone Quality in Chronic Kidney Disease: Definitions and Diagnostics. Curr. Osteoporos. Rep. 2017, 15, 207–213. [Google Scholar] [CrossRef]
- Dušejovská, M.; Vecka, M.; Rychlík, I.; Žák, A. Dyslipidemia in patients with chronic kidney disease: Etiology and management. Vnitrni Lekarstvi 2020, 66, 275–281. [Google Scholar] [CrossRef]
- Hamzaoui, M.; Djerada, Z.; Brunel, V.; Mulder, P.; Richard, V.; Bellien, J.; Guerrot, D. 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRj mice. Sci. Rep. 2020, 10, 1524. [Google Scholar] [CrossRef]
- Jia, T.; Olauson, H.; Lindberg, K.; Amin, R.; Edvardsson, K.; Lindholm, B.; Andersson, G.; Wernerson, A.; Sabbagh, Y.; Schiavi, S.; et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013, 14, 116–118. [Google Scholar] [CrossRef]
- Yamato, M.; Kato, N.; Kakino, A.; Yamada, K.I.; Inoguchi, T. Low dose of sodium-glucose transporter 2 inhibitor ipragliflozin attenuated renal dysfunction and intersti-tial fibrosis in adenine-induced chronic kidney disease in mice without diabetes. Metabol. Open 2020, 7, 100049. [Google Scholar] [CrossRef]
- Rahman, A.; Yamazaki, D.; Sufiun, A.; Kitada, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. A novel approach to adenine-induced chronic kidney disease associated anemia in rodents. PLoS ONE 2018, 13, e0192531. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, J.; Wolf, M. Regulation and Effects of FGF23 in Chronic Kidney Disease. Annu. Rev. Physiol. 2020, 82, 365–390. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Banga, R.S.; Kapitsinou, P.; Ramaiah, M.; Lawrence, J.; Kambhampati, G.; Gruenwald, A.; Bottinger, E.; Glicklich, D.; Tellis, V.; et al. Human and Murine Kidneys Show Gender- and Species-Specific Gene Expression Differences in Response to Injury. PLoS ONE 2009, 4, e4802. [Google Scholar] [CrossRef] [PubMed]
- Pecks, U.; Mohaupt, M.G.; Hütten, M.C.; Maass, N.; Rath, W.; Escher, G. Cholesterol acceptor capacity is preserved by different mechanisms in preterm and term fetuses. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 251–258. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Song, S.; Meyer, M.; Turk, T.R.; Wilde, B.; Feldkamp, T.; Assert, R.; Wu, K.; Kribben, A.; Witzke, O. Serum cystatin C in mouse models: A reliable and precise marker for renal function and superior to serum creatinine. Nephrol. Dial. Transplant. 2008, 24, 1157–1161. [Google Scholar] [CrossRef]
- Eisner, C.; Faulhaber-Walter, R.; Wang, Y.; Leelahavanichkul, A.; Yuen, P.; Mizel, D.; Star, R.A.; Briggs, J.; Levine, M.; Schnermann, J. Major contribution of tubular secretion to creatinine clearance in mice. Kidney Int. 2010, 77, 519–526. [Google Scholar] [CrossRef]
- Nakanishi, N.; Fukui, M.; Tanaka, M.; Toda, H.; Imai, S.; Yamazaki, M.; Hasegawa, G.; Oda, Y.; Nakamura, N. Low Urine pH Is a Predictor of Chronic Kidney Disease. Kidney Blood Press. Res. 2011, 35, 77–81. [Google Scholar] [CrossRef]
- Dos Santos, I.F.; Sheriff, S.; Amlal, S.; Ahmed, R.P.H.; Thakar, C.V.; Amlal, H. Adenine acts in the kidney as a signaling factor and causes salt- and water-losing nephropathy: Early mechanism of adenine-induced renal injury. Am. J. Physiol. Physiol. 2019, 316, 743–757. [Google Scholar] [CrossRef]
- Sakr, S.W.; Williams, D.L.; Stoudt, G.W.; Phillips, M.C.; Rothblat, G.H. Induction of cellular cholesterol efflux to lipid-free apolipoprotein A-I by cAMP. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 1999, 1438, 85–98. [Google Scholar] [CrossRef]
- Wang, N.; Westerterp, M. ABC Transporters, Cholesterol Efflux, and Implications for Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1276, 67–83. [Google Scholar] [PubMed]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116.1. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.; Madanagopal, T.; Ramkumar, A.; Boudaka, A.; Tageldin, M.; Nemmar, A. Some physiological and histological aspects of the gastrointestinal tract in a mouse model of chronic renal failure. J. Pharmacol. Toxicol. Methods 2014, 69, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Handa, Y.; Uematsu, Y.; Takeda, S.; Sekine, K.; Yoshihara, Y.; Kawakami, T.; Arioka, K.; Sato, H.; Uchiyama, Y.; et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat. Genet. 1997, 16, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Rowling, M.J.; Gliniak, C.; Welsh, J.; Fleet, J.C. High Dietary Vitamin D Prevents Hypocalcemia and Osteomalacia in CYP27B1 Knockout Mice12. J. Nutr. 2007, 137, 2608–2615. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NO | Adenine | p Value |
|---|---|---|---|
| n = 10 | n = 9 | ||
| URINE | |||
| Creatinine [µmol/24 h] | 3.33 ± 0.38 | 3.79 ± 0.27 | NS |
| Na+ [µmol/24 h] | 112.4 ± 13.9 | 175.4 ± 18.3 | 0.0125 |
| K+ [µmol/24 h] | 247.8 ± 34.05 | 308.4 ± 27.7 | NS |
| Ca2+ [µmol/24 h] | 1.01 ± 0.11 | 6.37 ± 2.07 | 0.0051 |
| Mg2+ [µmol/24 h] | 6.49 ± 0.71 | 12.54 ± 1.93 | 0.0032 |
| Pi [µmol/24 h] | 86.09 ± 11.87 | 102.1 ± 6.15 | 0.0236 |
| Cl− [µmol/24 h] | 132 ± 18.33 | 163.8 ± 17.8 | NS |
| Urea [µmol/24 h] | 1271 ± 150.5 | 1432 ± 105.2 | NS |
| Glucose [µmol/24 h] | 1.51 ± 0.155 | 1.98 ± 0.24 | NS |
| SERUM | |||
| Creatinine [µmol/L] | 36.6 ± 7.5 | 32.1 ± 9.0 | NS |
| BUN [mg/dL] | 39.28 ± 3.88 | 43.22 ± 5.32 | NS |
| Osmolality [mOsmol/kg] | 3492.0 ± 497.2 | 1026.0 ± 110.0 | 0.0003 |
| FGF23 [pg/mL] | 253.0 ± 92.3 | 1038.0 ± 403.5 | 0.0003 |
| Glucose [mmol/L] | 5.5 ± 0.26 | 6.6 ± 0.4 | 0.0272 |
| RENAL FUNCTION | |||
| UreaUrine/UreaSerum | 156.3 ± 23.4 | 42.3 ± 8.1 | 0.0008 |
| CreatinineUrine/CreatinineSerum | 338.6 ± 201.8 | 112.4 ± 47.3 | 0.0011 |
| NO | Adenine | p Value | |
|---|---|---|---|
| n = 10 | n = 9 | ||
| SERUM | |||
| Total Cholesterol [mg/dL] | 503.3 ± 41.6 | 619.1 ± 51.3 | NS |
| HDL [mg/mL] | 0.08 ± 0.01 | 0.08 ± 0.01 | NS |
| LDL [mg/mL] | 2.24 ± 0.14 | 2.63 ± 0.21 | NS |
| Triglycerides [mg/dL] | 74.58 ± 15.94 | 93.73 ± 15.09 | NS |
| HDL/LDL | 3.54 ± 0.62 | 3.29 ± 0.51 | NS |
| LIVER | |||
| Cholesterol [µg/mg] | 10.84 ± 0.73 | 9.74 ± 0.51 | NS |
| Cholesteryl ester [µg/mg] | 5.24 ± 0.81 | 4.79 ± 0.67 | NS |
| Trigylcerides [nmol/mg] | 14.64 ± 1.08 | 12.62 ± 1.27 | NS |
| FECES | |||
| Cholesterol [mg/24 h] | 3.92 ± 0.52 | 5.21 ± 0.8 | NS |
| Cholesteryl ester [mg/24 h] | 0.35 ± 0.05 | 0.67 ± 0.12 | 0.0197 |
| Trigylcerides [mg/24 h] | 0.035 ± 0.007 | 0.149 ± 0.033 | 0.0042 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scherler, L.; Verouti, S.N.; Ackermann, D.; Vogt, B.; Escher, G. Adenine-Induced Nephropathy Reduces Atherosclerosis in ApoE Knockout Mice. Biomolecules 2022, 12, 1147. https://doi.org/10.3390/biom12081147
Scherler L, Verouti SN, Ackermann D, Vogt B, Escher G. Adenine-Induced Nephropathy Reduces Atherosclerosis in ApoE Knockout Mice. Biomolecules. 2022; 12(8):1147. https://doi.org/10.3390/biom12081147
Chicago/Turabian StyleScherler, Laeticia, Sofia N. Verouti, Daniel Ackermann, Bruno Vogt, and Geneviève Escher. 2022. "Adenine-Induced Nephropathy Reduces Atherosclerosis in ApoE Knockout Mice" Biomolecules 12, no. 8: 1147. https://doi.org/10.3390/biom12081147
APA StyleScherler, L., Verouti, S. N., Ackermann, D., Vogt, B., & Escher, G. (2022). Adenine-Induced Nephropathy Reduces Atherosclerosis in ApoE Knockout Mice. Biomolecules, 12(8), 1147. https://doi.org/10.3390/biom12081147