
Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease



Abstract
:1. Introduction
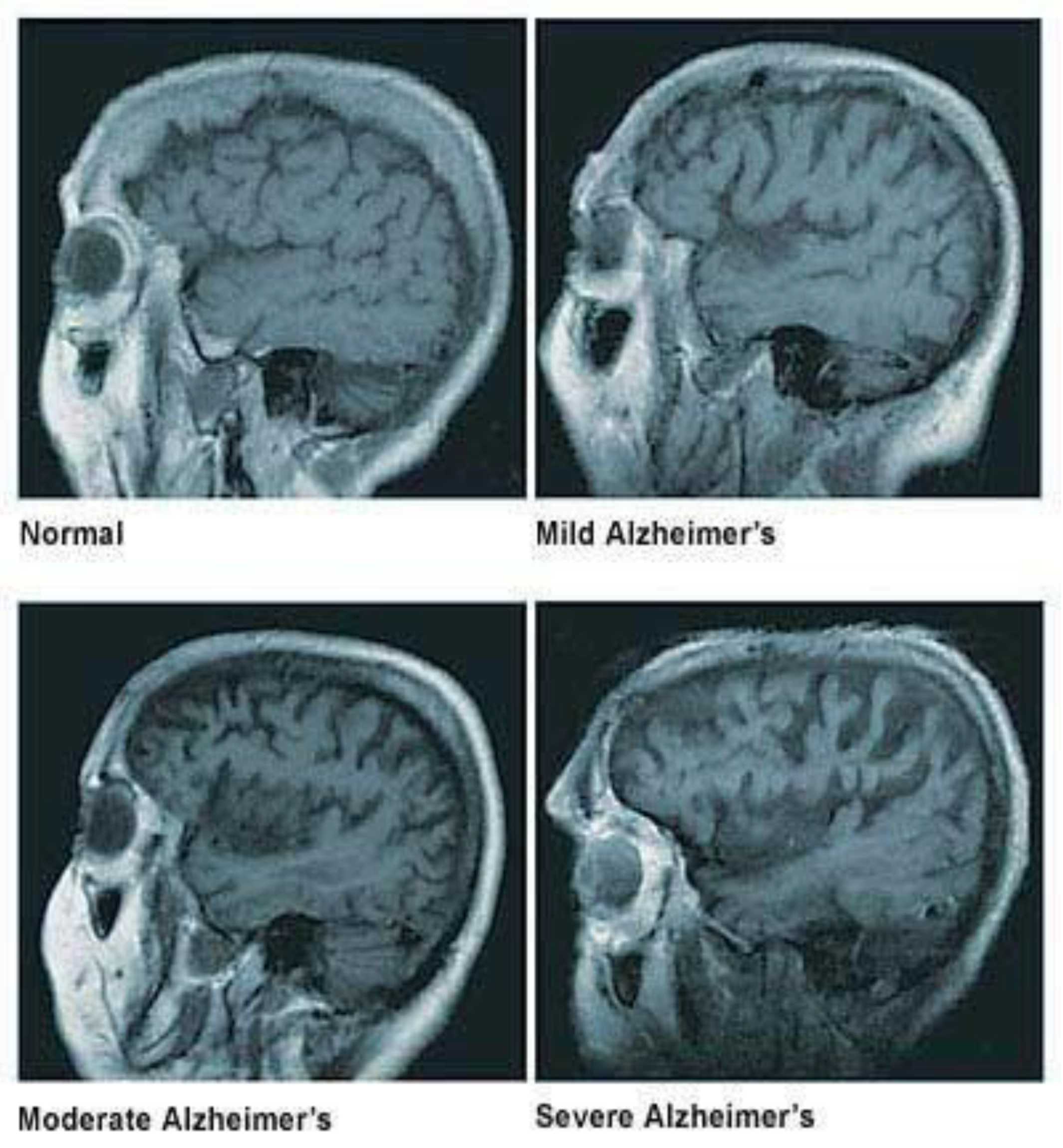
1.1. Loss of Neuronal Connections & Cell Death in AD
1.2. Causes & Diagnosis of AD
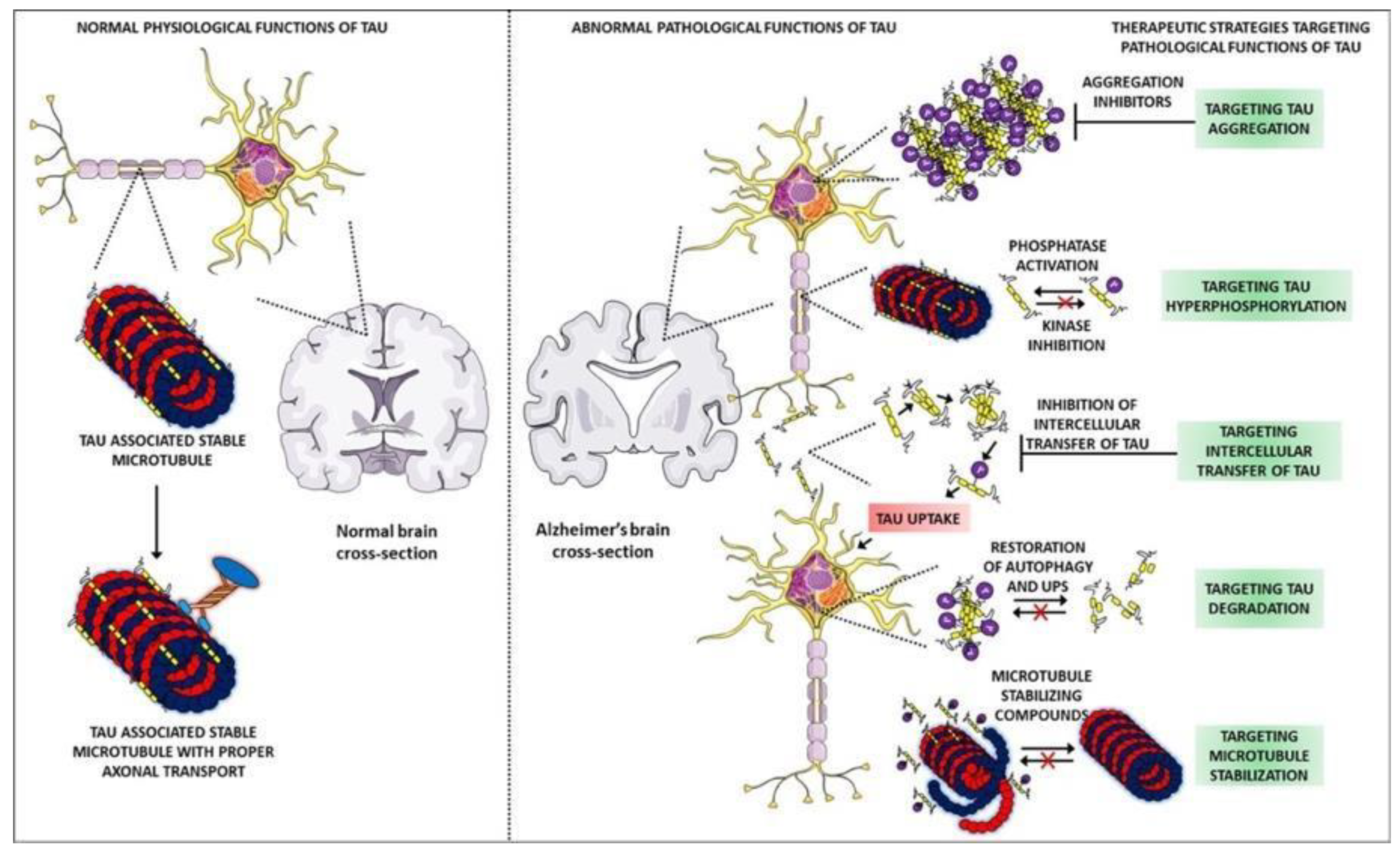
1.3. Description of Tau Protein and Its Mechanisms
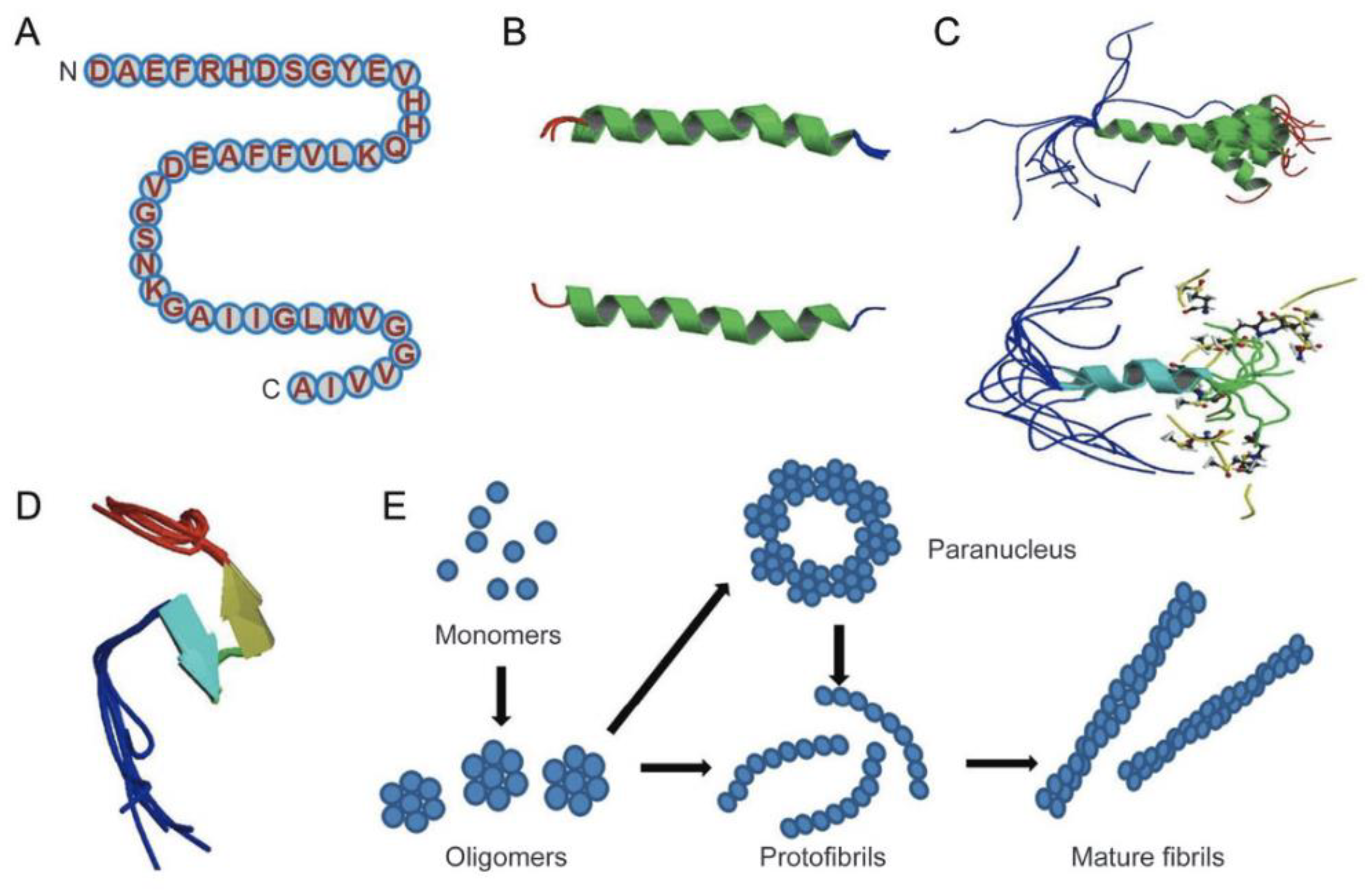
1.4. Structure of Amyloid-β Peptide
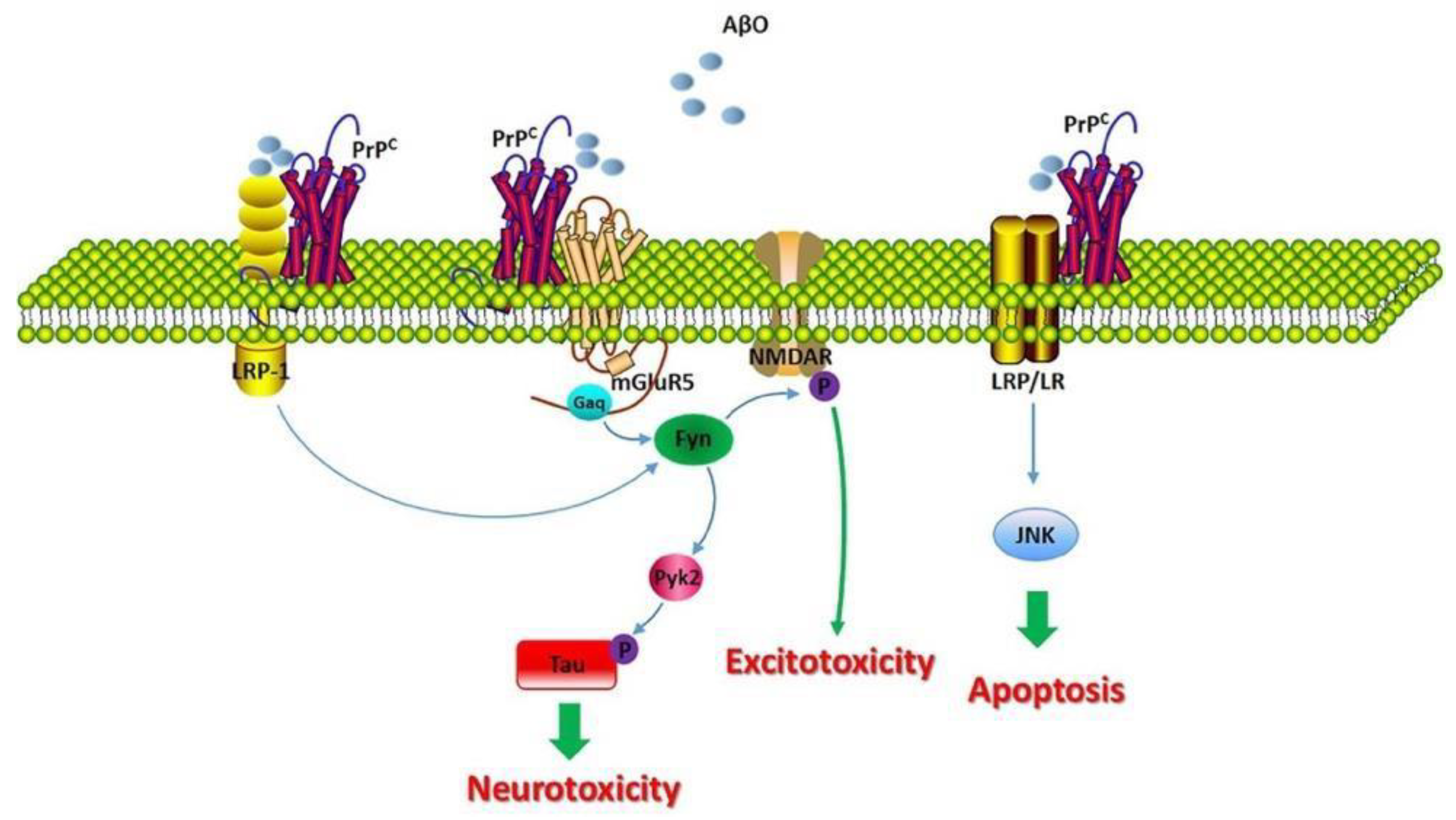
1.5. Description of Amyloid-β Protein and Its Mechanisms
1.6. Effects of Acetylcholinergic Neurons
1.7. Targeting in AD
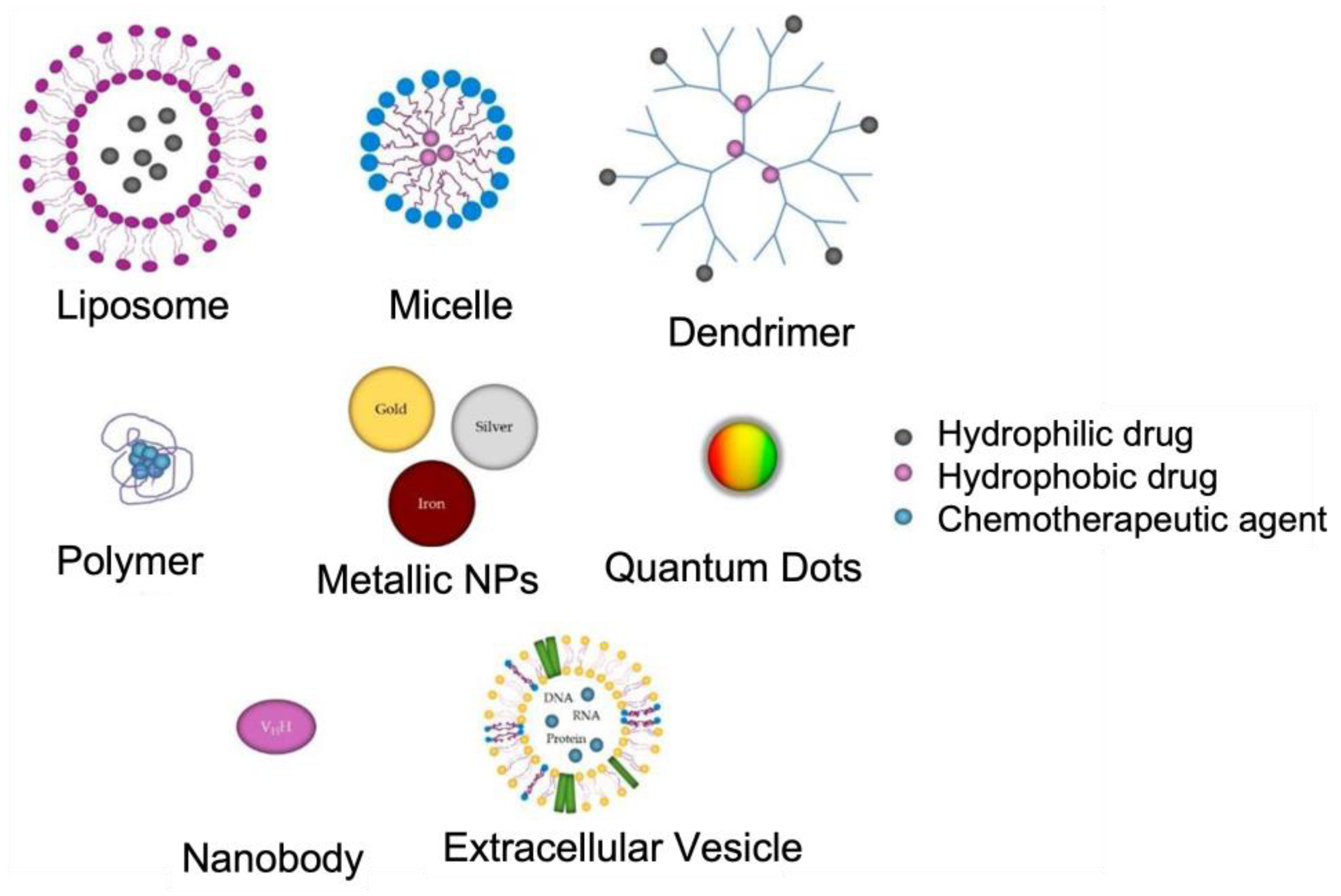
2. Nanotechnology for AD
3. Polymeric NPs in the Treatment of AD
3.1. Polymeric Porous Nanomaterial for AD Biomarker Analysis
3.2. Polymeric NPs in Tau Targeted Immunotherapy
3.3. Polymeric NPs in Amyloid-β Targeted Immunotherapy
4. Liposome NPs
4.1. Liposome NPs in Tau Targeted Immunotherapy
4.2. Liposome NPs in Amyloid-β Targeted Immunotherapy
5. Metallic NPs
5.1. Metallic NPs in Tau Targeted Immunotherapy
5.2. Metallic NPs in Amyloid-β Targeted Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Carrier | Composition | Size (nm) | Route of Admin. | Ligand BBB | Ligand AD Target | Drug Loaded | Function | Reference |
|---|---|---|---|---|---|---|---|---|
| Polymeric | DGL-PEG | 95–110 | I.v. | Peptide (RVG29) | Peptide (RVG29) | pshBACE1-AS, Peptide (D-Pep) | Downregulation of BACE1 level inhibition of phosphorylated- tau related fibril | [29] |
| Polymeric (micelles) | hydrophobic poly(ε-caprolacton), hydrophilic poly (ethylene glycol) | 33 | In vitro | - | Peptide (D)-TLKIVW | Tau-targeted multifunctional inhibitor | [30] | |
| Liposome | DMPC, DMPG, CH, MLPA | S.c. | - | - | Peptide (tau-fragment) | Tau immunotherapy | [36] | |
| Liposome | RBCm-coated | 150–200 | I.v. | T807 | Curcumin | CUR-loaded T807/RPCNP NPs | Tau targeting | [37] |
| Metallic | protein-capped | 50–60 | In vitro | - | - | 10% SDS-PAGE | Tau aggregation inhibitor | [42] |
| Metallic (gold) | poly(amidoamine) (PAMAM) dendrimer nanocomposite (3D-Au-PAMAM) | 2–100 | Antibody (CAb), antibody (HRP-DAb) | - | Direct determination of tau | [43] | ||
| Carbon-based | PX@OMCN–PEG, PX@OP@RVG | 110 | In vivo and in vitro | RVG peptide | RVG peptide | Protoporphyrin IX (PX) | Tau phosphorylation inhibitor | [52] |
| Type of Carrier | Composition | Size (nm) | Route of Admin. | Ligand BBB | Ligand AD Target | Drug Loaded | Function | Reference |
|---|---|---|---|---|---|---|---|---|
| Liposome | DPPC, CH, DPS-curcumin. | 200 | I.c.v. | - | Curcumin | - | Targeting Aβ | [38,39] |
| Liposome | DPPC, CH, DSPE-PEG | 150 | I.v. | Passive targeting | Methoxy XO4 | - | Ttargeting Aβ | [40] |
| Liposome | PC or DSPC, CH, DSPE- PEG | 140–170 | In vitro | Antibody (OX26Mab) | Antibody (AβMab) | - | Targeting Aβ | [46] |
| Liposome | SM, CH, DMPA, DSPE- PEG | 130 | In vitro | Antibody (RI7217) | PA | - | Targeting Aβ | [47] |
| Liposome | DSPC, CH, DSPE-PEG | 90–120 | I.v. | Antibody (OX26Mab) | Antibody (19B8MAb) | - | Targeting Aβ | [48] |
| Liposome | SM, CH, DMPA | 100–150 | I.p. | Peptide (mApoE) | PA | - | Destabilize brain Aβ aggregates and promote peptide removal | [49] |
| Liposome | DMPC | 20–35 | I.v. | Protein (ApoE3) | Protein (ApoE3) | - | Decreased amyloid deposition | [50] |
| Liposome | PC, DSPE- PEG | 80–300 | I.v. | Protein (lactoferrin) | - | Peptide (KLVFF) and curcumin | β-sheet breaker antioxidant | [51] |
| Polymeric | PEG-PLA | 90–110 | I.v. | Peptide (TGN) | D-Peptide (D-QSH) | Peptide (H102) | [31] | |
| Metallic (gold) | Au | 20–25 | I.v. | Passive | Peptide (LPFFD) | POMD | Inhibition of aggregation Dissociation antioxidant | [44] |
| Metallic (gold) | Au | 15 | I.v. | Peptide (THR) | Peptide (LPFFD) | - | Inhibition of aggregation | [45] |
6. Conclusions
6.1. Comparison between Tau and Amyloid-β
6.2. Future Outlook with Nanotechnology, Molecular Modeling and Simulation
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institutes of Health. National Institute on Aging What Happens to the Brain in Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease (accessed on 25 May 2022).
- Pini, L.; Pievani, M.; Bocchetta, M.; Altomare, D.; Bosco, P.; Cavedo, E.; Galluzzi, S.; Marizzoni, M.; Frisoni, G.B. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res. Rev. 2016, 30, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Illenberger, S.; Zheng-Fischhöfer, Q.; Preuss, U.; Stamer, K.; Baumann, K.; Trinczek, B.; Biernat, J.; Godemann, R.; Mandelkow, E.M.; Mandelkow, E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol. Biol. Cell 1998, 9, 1495–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of Tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Theoni Gyparaki, M.; Arab, A.; Sorokina, E.M.; Santiago-Ruiz, A.N.; Bohrer, C.H.; Xiao, J.; Lakadamyali, M. Tau Forms Oligomeric Complexes on Microtubules that are Distinct from Pathological Oligomers in Disease. Biophys. J. 2021, 120, 301a. [Google Scholar] [CrossRef]
- Gorlovoy, P.; Larionov, S.; Pham, T.T.H.; Neumann, H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J. 2009, 23, 2502–2513. [Google Scholar] [CrossRef]
- Robertson, L.A.; Moya, K.L.; Breen, K.C. The potential role of tau protein O-glycosylation in Alzheimer’s disease. J. Alzheimer’s Dis. 2004, 6, 489–495. [Google Scholar] [CrossRef]
- Cohen, T.J.; Constance, B.H.; Hwang, A.W.; James, M.; Yuan, C.-X. Intrinsic Tau Acetylation Is Coupled to Auto-Proteolytic Tau Fragmentation. PLoS ONE 2016, 11, e0158470. [Google Scholar] [CrossRef]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.-X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Gallardo, G.; Wong, C.H.; Ricardez, S.M.; Mann, C.N.; Lin, K.H.; Leyns, C.E.G.; Jiang, H.; Holtzman, D.M. Targeting tauopathy with engineered tau-degrading intrabodies. Mol. Neurodegener. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Gao, C.; Chu, X.; Gong, W.; Zheng, J.; Xie, X.; Wang, Y.; Yang, M.; Li, Z.; Gao, C.; Yang, Y. Neuron tau-targeting biomimetic nanoparticles for curcumin delivery to delay progression of Alzheimer’s disease. J. Nanobiotechnol. 2020, 18, 71. [Google Scholar] [CrossRef]
- Macleod, R.; Hillert, E.K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer’s disease. Futur. Sci. OA 2015, 1, FSO11. [Google Scholar] [CrossRef] [Green Version]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Zhang, L.; Yu, W.; Wang, Y.; Chang, W. Cellular Prion Protein as a Receptor of Toxic Amyloid-β42 Oligomers Is Important for Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 339. [Google Scholar] [CrossRef] [Green Version]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2, 336. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S67–S78. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Mouri, A.; Noda, Y.; Hara, H.; Mizoguchi, H.; Tabira, T.; Nabeshima, T. Oral vaccination with a viral vector containing Aβ cDNA attenuates age-related Aβ accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J. 2007, 21, 2135–3148. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.K.; Park, S.Y. Neuroprotective effect of cellular prion protein (PrPC) is related with activation of alpha7 nicotinic acetylcholine receptor (a7nAchR)-mediated autophagy flux. Oncotarget 2015, 6, 24660–24674. [Google Scholar] [CrossRef]
- Amit, T.; Avramovich-Tirosh, Y.; Youdim, M.B.H.; Mandel, S. Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008, 22, 1296–1305. [Google Scholar] [CrossRef] [Green Version]
- Olazarán, J.; Reisberg, B.; Clare, L.; Cruz, I.; Peña-Casanova, J.; Del Ser, T.; Woods, B.; Beck, C.; Auer, S.; Lai, C.; et al. Nonpharmacological therapies in alzheimer’s disease: A systematic review of efficacy. Dement. Geriatr. Cogn. Disord. 2010, 30, 161–178. [Google Scholar] [CrossRef]
- Khan, N.H.; Mir, M.; Ngowi, E.E.; Zafar, U.; Khakwani, M.M.A.K.; Khattak, S.; Zhai, Y.K.; Jiang, E.S.; Zheng, M.; Duan, S.F.; et al. Nanomedicine: A Promising Way to Manage Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2021, 9, 630055. [Google Scholar] [CrossRef]
- Martín-Rapun, R.; De Matteis, L.; Ambrosone, A.; Garcia-Embid, S.; Gutierrez, L.; de la Fuente, J.M. Targeted Nanoparticles for the Treatment of Alzheimer’s Disease. Curr. Pharm. Des. 2017, 23, 1927–1952. [Google Scholar] [CrossRef] [Green Version]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Mahmoodi, N.O.; Ghavidast, A.; Amirmahani, N. A comparative study on the nanoparticles for improved drug delivery systems. J. Photochem. Photobiol. B Biol. 2016, 162, 681–693. [Google Scholar] [CrossRef]
- Alja, Z.; Alja Videtic Paska, I.J. Nanotechnology Meets Oncology: Nanomaterials in brain cancer research, diagnosis and therap. Materials 2019, 12, 1588. [Google Scholar]
- Husseini, G.A.; Pitt, W.G. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1137–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carradori, D.; Balducci, C.; Re, F.; Brambilla, D.; Le Droumaguet, B.; Flores, O.; Gaudin, A.; Mura, S.; Forloni, G.; Ordoñez-Gutierrez, L.; et al. Antibody-functionalized polymer nanoparticle leading to memory recovery in Alzheimer’s disease-like transgenic mouse model. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, W.; Lu, Y.; Xu, Y.; Wang, C.; Yu, D.-G.; Kim, I. Recent Advances in Poly(α-L-glutamic acid)-Based Nanomaterials for Drug Delivery. Biomolecules 2022, 12, 636. [Google Scholar] [CrossRef]
- Liu, Y.; An, S.; Li, J.; Kuang, Y.; He, X.; Guo, Y.; Ma, H.; Zhang, Y.; Ji, B.; Jiang, C. Brain-targeted co-delivery of therapeutic gene and peptide by multifunctional nanoparticles in Alzheimer’s disease mice. Biomaterials 2016, 80, 33–45. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, L.; Wu, X.; Deng, F.; Ma, R.; Liu, Y.; Huang, F.; Shi, L. Tau-Targeted Multifunctional Nanoinhibitor for Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2021, 13, 23328–23338. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, X.; Wan, X.; Shao, X.; Liu, Q.; Zhang, Z.; Zhang, Q. The potential use of H102 peptide-loaded dual-functional nanoparticles in the treatment of Alzheimer’s disease. J. Control. Release 2014, 192, 317–324. [Google Scholar] [CrossRef]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef]
- Micheli, M.R.; Bova, R.; Magini, A.; Polidoro, M.; Emiliani, C. Lipid-based nanocarriers for CNS-targeted drug delivery. Recent Pat. CNS Drug Discov. 2012, 7, 71–86. [Google Scholar] [CrossRef]
- Wong, H.L.; Wu, X.Y.; Bendayan, R. Nanotechnological advances for the delivery of CNS therapeutics. Adv. Drug Deliv. Rev. 2012, 64, 686–700. [Google Scholar] [CrossRef]
- Rip, J.; Chen, L.; Hartman, R.; Van Den Heuvel, A.; Reijerkerk, A.; Van Kregten, J.; Van Der Boom, B.; Appeldoorn, C.; De Boer, M.; Maussang, D.; et al. Glutathione PEGylated liposomes: Pharmacokinetics and delivery of cargo across the blood-brain barrier in rats. J. Drug Target. 2014, 22, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in Tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [Green Version]
- Lazar, A.N.; Mourtas, S.; Youssef, I.; Parizot, C.; Dauphin, A.; Delatour, B.; Antimisiaris, S.G.; Duyckaerts, C. Curcumin-conjugated nanoliposomes with high affinity for Aβ deposits: Possible applications to Alzheimer disease. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 712–721. [Google Scholar] [CrossRef]
- Fan, S.; Zheng, Y.; Liu, X.; Fang, W.; Chena, X.; Liao, W.; Jing, X.; Lei, M.; Tao, E.; Ma, Q.; et al. Curcumin-loaded plga-peg nanoparticles conjugated with b6 peptide for potential use in alzheimer’s disease. Drug Deliv. 2018, 25, 1091–1102. [Google Scholar] [CrossRef] [Green Version]
- Tanifum, E.A.; Dasgupta, I.; Srivastava, M.; Bhavane, R.C.; Sun, L.; Berridge, J.; Pourgarzham, H.; Kamath, R.; Espinosa, G.; Cook, S.C.; et al. Intravenous Delivery of Targeted Liposomes to Amyloid-β Pathology in APP/PSEN1 Transgenic Mice. PLoS ONE 2012, 7, e48515. [Google Scholar] [CrossRef]
- Singh, P.; Pandit, S.; Mokkapati, V.R.S.S.; Garg, A.; Ravikumar, V.; Mijakovic, I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int. J. Mol. Sci. 2018, 19, 197. [Google Scholar] [CrossRef]
- Sonawane, S.K.; Ahmad, A.; Chinnathambi, S. Protein-Capped Metal Nanoparticles Inhibit Tau Aggregation in Alzheimer’s Disease. ACS Omega 2019, 4, 12833–12840. [Google Scholar] [CrossRef] [Green Version]
- Razzino, C.A.; Serafín, V.; Gamella, M.; Pedrero, M.; Montero-Calle, A.; Barderas, R.; Calero, M.; Lobo, A.O.; Yáñez-Sedeño, P.; Campuzano, S.; et al. An electrochemical immunosensor using gold nanoparticles-PAMAM-nanostructured screen-printed carbon electrodes for tau protein determination in plasma and brain tissues from Alzheimer patients. Biosens. Bioelectron. 2020, 163, 112238. [Google Scholar] [CrossRef]
- Gao, N.; Sun, H.; Dong, K.; Ren, J.; Qu, X. Gold-nanoparticle-based multifunctional amyloid-β inhibitor against Alzheimer’s disease. Chemistry 2015, 21, 829–835. [Google Scholar] [CrossRef]
- Prades, R.; Guerrero, S.; Araya, E.; Molina, C.; Salas, E.; Zurita, E.; Selva, J.; Egea, G.; López-Iglesias, C.; Teixidó, M.; et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials 2012, 33, 7194–7205. [Google Scholar] [CrossRef]
- Markoutsa, E.; Papadia, K.; Clemente, C.; Flores, O.; Antimisiaris, S.G. Anti-Aβ-MAb and dually decorated nanoliposomes: Effect of Aβ1-42 peptides on interaction with hCMEC/D3 cells. Eur. J. Pharm. Biopharm. 2012, 81, 49–56. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashal, Y.; Abdelhady, H.; Iyer, A.K. Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease. Biomolecules 2022, 12, 1001. https://doi.org/10.3390/biom12071001
Mashal Y, Abdelhady H, Iyer AK. Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease. Biomolecules. 2022; 12(7):1001. https://doi.org/10.3390/biom12071001
Chicago/Turabian StyleMashal, Yara, Hosam Abdelhady, and Arun K. Iyer. 2022. "Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease" Biomolecules 12, no. 7: 1001. https://doi.org/10.3390/biom12071001
APA StyleMashal, Y., Abdelhady, H., & Iyer, A. K. (2022). Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease. Biomolecules, 12(7), 1001. https://doi.org/10.3390/biom12071001