
Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy

Abstract
:1. Introduction
2. Overview of FEN1
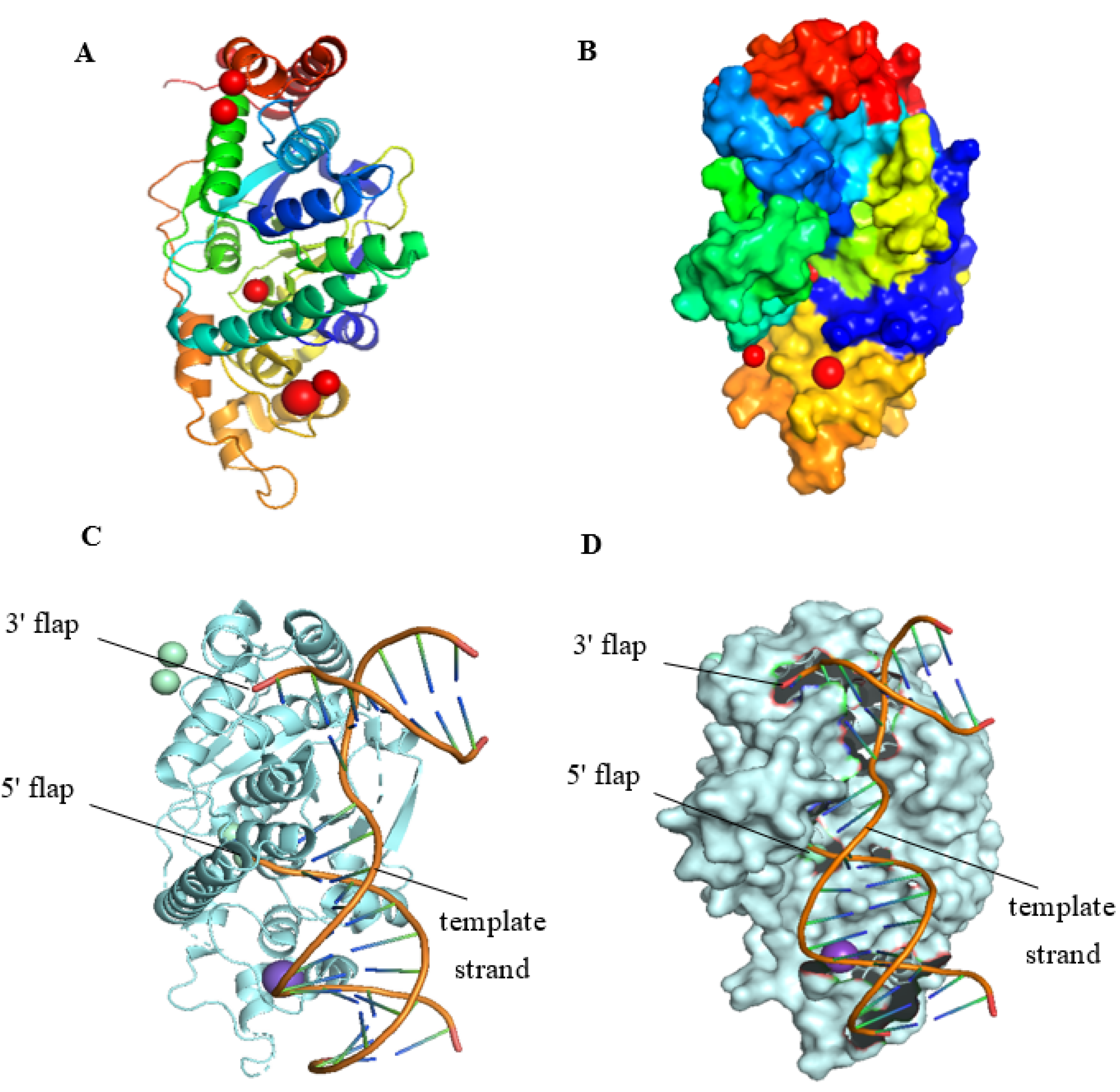
2.1. The Structure of FEN1
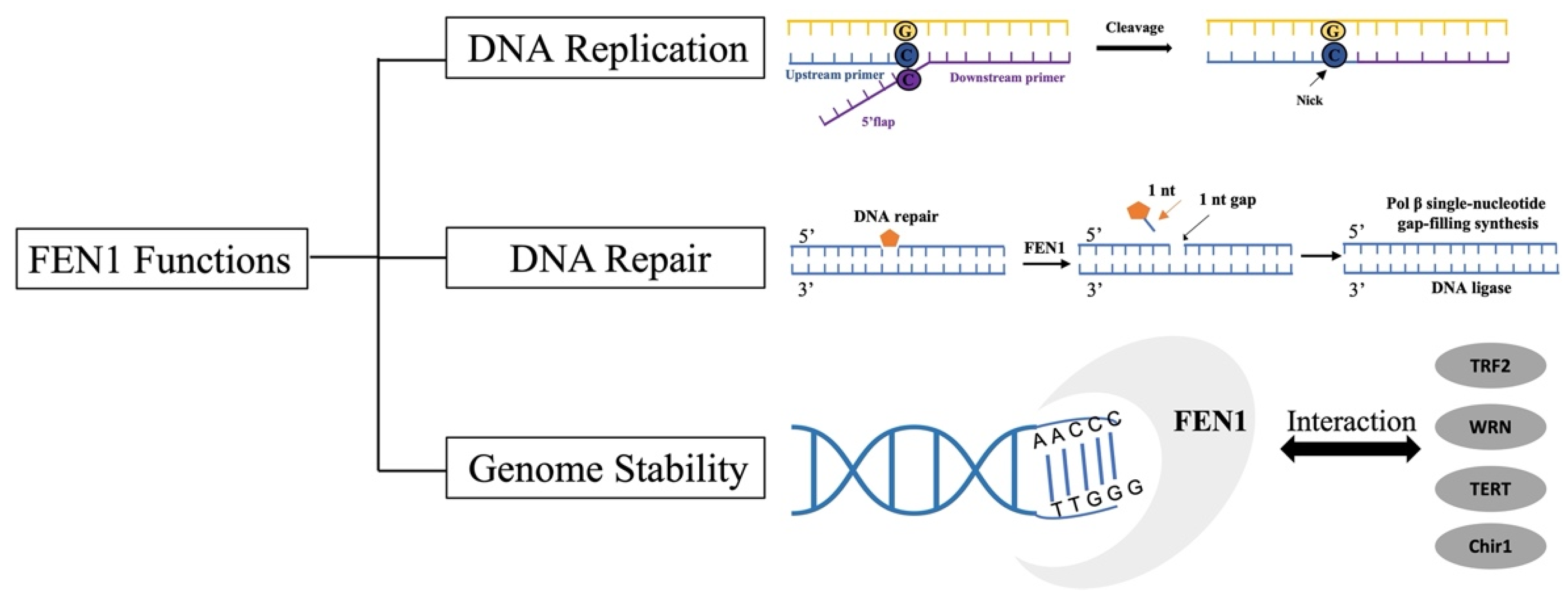
2.2. FEN1 Functions
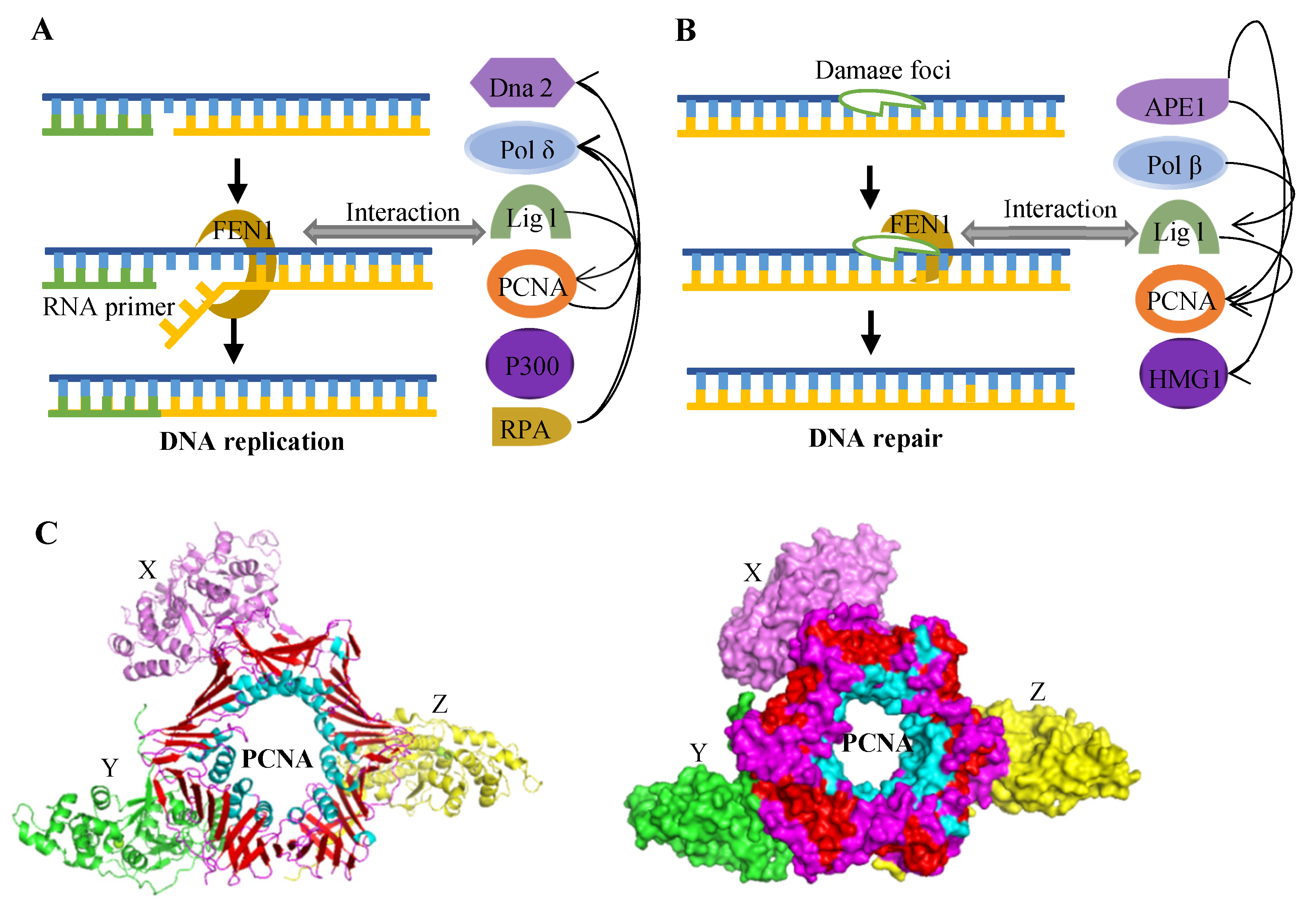
2.2.1. Okazaki Fragment Maturation
2.2.2. DNA BERP Pathway
2.2.3. Other DNA Repair Pathways
2.2.4. Genome Stability
2.2.5. Telomere Stability
2.3. Post-Transcriptional Regulation Mechanism of FEN1
2.4. Proteins Interacting with FEN1
3. The Role of FEN1 in Diseases
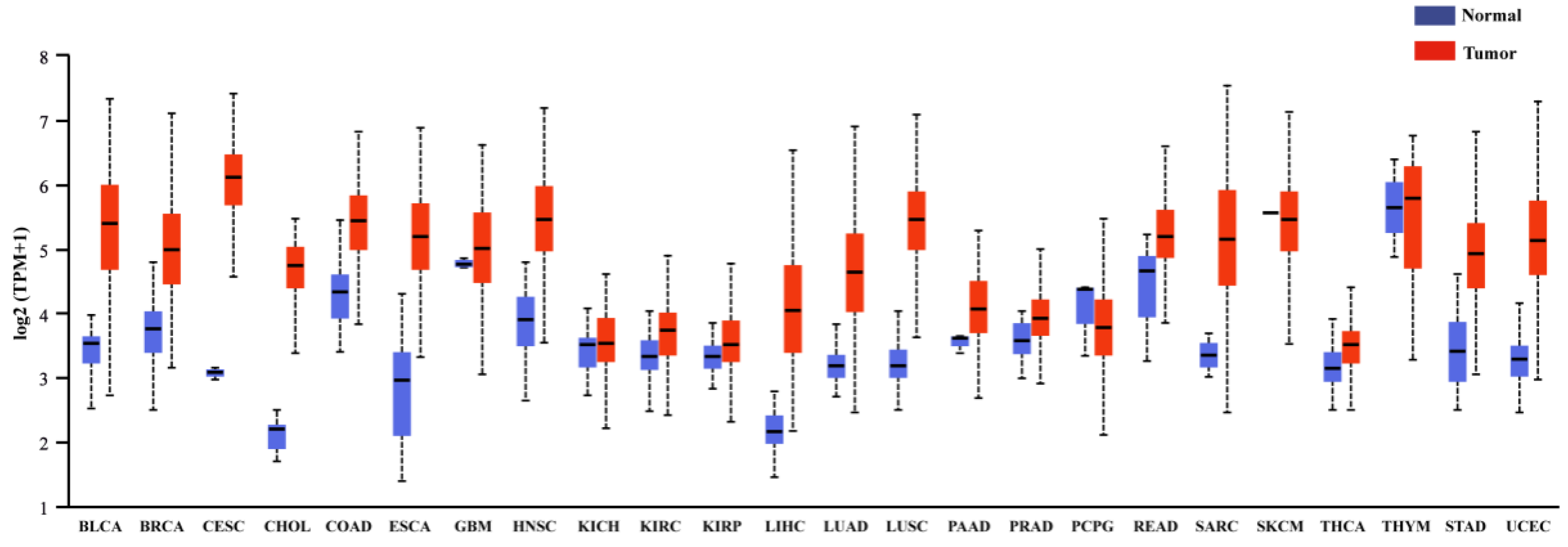
3.1. Cancers
3.2. Other Diseases
4. Development of FEN1 Inhibitors
4.1. Structural Design of Small-Molecule Inhibitors
4.2. Small-Molecule Inhibitors Targeting FEN1
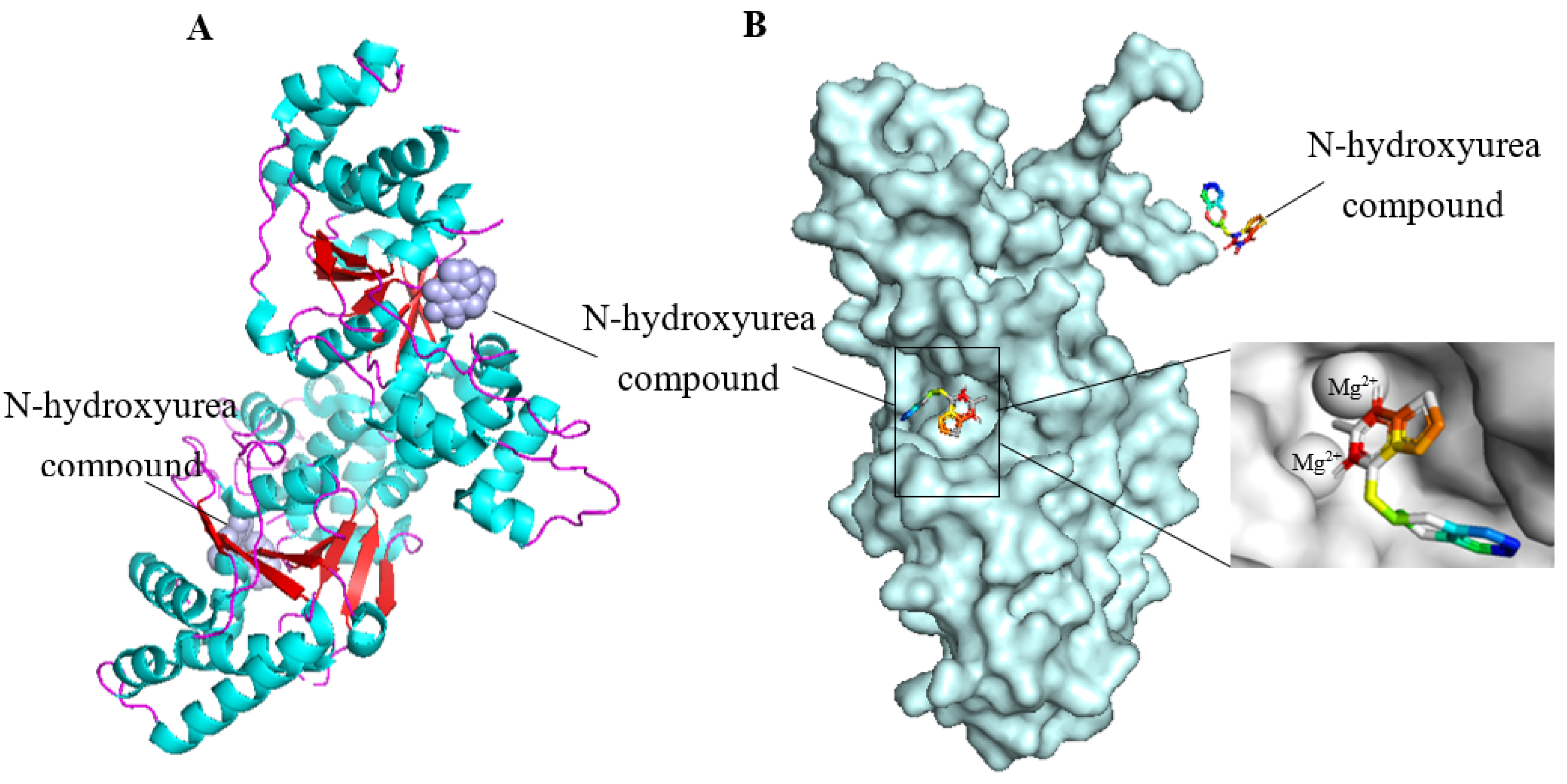
4.2.1. FEN1 Inhibitors of N-hydroxy Urea
4.2.2. FEN1i
4.2.3. FEN1i #2
4.2.4. SC13
4.2.5. Other Inhibitors
4.3. Defects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022, 36, 278–293. [Google Scholar] [CrossRef]
- Hosfield, D.J.; Mol, C.D.; Shen, B.; Tainer, J.A. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: Coupling DNA and PCNA binding to FEN-1 activity. Cell 1998, 95, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Jia, J.; Finger, L.D.; Guo, Z.; Zer, C.; Shen, B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011, 39, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Prasad, R.; Beard, W.A.; Hou, E.W.; Horton, J.K.; McMurray, C.T.; Wilson, S.H. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 2009, 284, 28352–28366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Zheng, L.; Chavez, V.; Qiu, J.; Shen, B. Concerted action of exonuclease and Gap-dependent endonuclease activities of FEN-1 contributes to the resolution of triplet repeat sequences (CTG)n- and (GAA)n-derived secondary structures formed during maturation of Okazaki fragments. J. Biol. Chem. 2007, 282, 3465–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saharia, A.; Guittat, L.; Crocker, S.; Lim, A.; Steffen, M.; Kulkarni, S.; Stewart, S.A. Flap endonuclease 1 contributes to telomere stability. Curr. Biol. 2008, 18, 496–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Dai, H.; Hegde, M.L.; Zhou, M.; Guo, Z.; Wu, X.; Wu, J.; Su, L.; Zhong, X.; Mitra, S.; et al. Fen1 mutations that specifically disrupt its interaction with PCNA cause aneuploidy-associated cancer. Cell Res. 2011, 21, 1052–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, G.; Wang, Y.; Wei, H.; Chen, M.; Lin, H.; Huang, Z.; Huang, J.; Wang, S.; Lin, J. The Clinical Significance of the Expression of FEN1 in Primary Osteosarcoma. Int. J. Gen. Med. 2021, 14, 6477–6485. [Google Scholar] [CrossRef]
- Mesquita, K.A.; Ali, R.; Doherty, R.; Toss, M.S.; Miligy, I.; Alblihy, A.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M.; et al. FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers 2021, 13, 1866. [Google Scholar] [CrossRef]
- Lu, X.; Liu, R.; Wang, M.; Kumar, A.K.; Pan, F.; He, L.; Hu, Z.; Guo, Z. MicroRNA-140 impedes DNA repair by targeting FEN1 and enhances chemotherapeutic response in breast cancer. Oncogene 2020, 39, 234–247. [Google Scholar] [CrossRef]
- He, L.; Zhang, Y.; Sun, H.; Jiang, F.; Yang, H.; Wu, H.; Zhou, T.; Hu, S.; Kathera, C.S.; Wang, X.; et al. Targeting DNA Flap endonuclease 1 to impede breast cancer progression. EBioMedicine 2016, 14, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhou, M.; Chai, Q.; Parrish, J.; Xue, D.; Patrick, S.M.; Turchi, J.J.; Yannone, S.M.; Chen, D.; Shen, B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005, 6, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kao, H.-I.; Bambara, R.A. Flap endonuclease 1: A central component of DNA metabolism. Annu. Rev. Biochem. 2004, 73, 589–615. [Google Scholar] [CrossRef]
- Xu, Y.; Grindley, N.D.; Joyce, C.M. Coordination between the polymerase and 5'-nuclease components of DNA polymerase I of Escherichia coli. J. Biol. Chem. 2000, 275, 20949–20955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutakawa, S.E.; Classen, S.; Chapados, B.R.; Arvai, A.S.; Finger, L.D.; Guenther, G.; Tomlinson, C.G.; Thompson, P.; Sarker, A.H.; Shen, B.; et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 2011, 145, 198–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, L.; Bambara, R.A. Flap endonuclease 1. Annu. Rev. Biochem. 2013, 82, 119–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Shi, R.; Han, W.; Cheng, J.; Xu, X.; Cheng, K.; Wang, L.; Tian, B.; Zheng, L.; Shen, B.; et al. Structural basis of 5'flap recognition and protein-protein interactions of human flap endonuclease 1. Nucleic Acids Res. 2018, 46, 11315–11325. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell. 2018, 71, 319–331.e3. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; He, L.; Wu, H.; Pan, F.; Wu, X.; Zhao, J.; Hu, Z.; Sekhar, C.; Li, H.; Zheng, L.; et al. The FEN1 L209P mutation interferes with long-patch base excision repair and induces cellular transformation. Oncogene 2017, 36, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Kleppa, L.; Mari, P.-O.; Larsen, E.; Lien, G.F.; Godon, C.; Theil, A.F.; Nesse, G.J.; Wiksen, H.; Vermeulen, W.; Giglia-Mari, G.; et al. Kinetics of endogenous mouse FEN1 in base excision repair. Nucleic Acids Res. 2012, 40, 9044–9059. [Google Scholar] [CrossRef] [Green Version]
- Ji, F.; Zhang, F.; Zhang, M.; Long, K.; Xia, M.; Lu, F.; Li, E.; Chen, J.; Li, J.; Chen, Z.; et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J. Hematol. Oncol. 2021, 14, 152. [Google Scholar] [CrossRef]
- Shen, M.; Young, A.; Autexier, C. PCNA, a focus on replication stress and the alternative lengthening of telomeres pathway. DNA Repair. 2021, 100, 103055. [Google Scholar] [CrossRef]
- Tom, S.; Henricksen, L.A.; Bambara, R.A. Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. J. Biol. Chem. 2000, 275, 10498–10505. [Google Scholar] [CrossRef] [Green Version]
- Thu, H.P.T.; Nguyen, T.A.; Munashingha, P.R.; Kwon, B.; Dao Van, Q.; Seo, Y.-S. A physiological significance of the functional interaction between Mus81 and Rad27 in homologous recombination repair. Nucleic Acids Res. 2015, 43, 1684–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, H.-M.; Tomkinson, A.E. Processing and joining of DNA ends coordinated by interactions among Dnl4/Lif1, Pol4, and FEN-1. J. Biol. Chem. 2004, 279, 47580–47588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutakawa, S.E.; Thompson, M.J.; Arvai, A.S.; Neil, A.J.; Shaw, S.J.; Algasaier, S.I.; Kim, J.C.; Finger, L.D.; Jardine, E.; Gotham, V.J.B.; et al. Phosphate steering by Flap Endonuclease 1 promotes 5'-flap specificity and incision to prevent genome instability. Nat. Commun. 2017, 8, 15855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, I.C.; Chen, B.C.; Shuai, H.-H.; Chien, F.-C.; Chen, P.; Hsieh, T.-S. Wuho is a new member in maintaining genome stability through its interaction with flap endonuclease 1. PLoS Biol. 2016, 14, e1002349. [Google Scholar] [CrossRef] [PubMed]
- Kucherlapati, M.; Nguyen, A.; Kuraguchi, M.; Yang, K.; Fan, K.; Bronson, R.; Wei, K.; Lipkin, M.; Edelmann, W.; Kucherlapati, R. Tumor progression in Apc(1638N) mice with Exo1 and Fen1 deficiencies. Oncogene 2007, 26, 6297–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Lu, Z.; Singh, A.; Zhou, Y.; Zheng, E.; Zhou, M.; Wang, J.; Wu, X.; Hu, Z.; Gu, Z.; et al. Error-prone, stress-induced 3'flap-based Okazaki fragment maturation supports cell survival. Science 2021, 374, 1252–1258. [Google Scholar] [CrossRef]
- Sampathi, S.; Bhusari, A.; Shen, B.; Chai, W. Human flap endonuclease I is in complex with telomerase and is required for telomerase-mediated telomere maintenance. J. Biol. Chem. 2009, 284, 3682–3690. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sommers, J.A.; Gary, R.K.; Friedrich-Heineken, E.; Hübscher, U.; Brosh, R.M. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005, 33, 6769–6781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Kanjanapangka, J.; Liu, N.; Liu, S.; Liu, C.; Wu, Z.; Wang, Y.; Loh, T.; Kowolik, C.; Jamsen, J.; et al. Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol. Cell 2012, 47, 444–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich-Heineken, E.; Hübscher, U. The Fen1 extrahelical 3'-flap pocket is conserved from archaea to human and regulates DNA substrate specificity. Nucleic Acids Res. 2004, 32, 2520–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zheng, L.; Xu, H.; Dai, H.; Zhou, M.; Pascua, M.R.; Chen, Q.M.; Shen, B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 2010, 6, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Shen, W.-J.; Kraemer, F.B.; Azhar, S. Regulation of adrenal and ovarian steroidogenesis by miR-132. J. Mol. Endocrinol. 2017, 59, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Luo, S. MiR-140-5p inhibits cervical cancer cell phenotypes via downregulating FEN1 to halt the cell cycle. Mol. Med. Rep. 2020, 22, 4919–4930. [Google Scholar] [CrossRef]
- Zhang, J.; Jing, L.; Tan, S.; Zeng, E.-M.; Lin, Y.; He, L.; Hu, Z.; Liu, J.; Guo, Z. Inhibition of miR-1193 leads to synthetic lethality in glioblastoma multiforme cells deficient of DNA-PKcs. Cell Death Dis. 2020, 11, 602. [Google Scholar] [CrossRef]
- Levikova, M.; Cejka, P. The Saccharomyces cerevisiae Dna2 can function as a sole nuclease in the processing of Okazaki fragments in DNA replication. Nucleic Acids Res. 2015, 43, 7888–7897. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, L.; Stewart, J.; Polaczek, P.; Campbell, J.L.; Bambara, R.A. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J. Biol. Chem. 2010, 285, 4398–4404. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, M.; Pang, Y.; Cheng, M.; Huang, B.; Xu, S.; Liu, M.; Lian, H.; Zhong, C. Flap endonuclease 1 and DNA-PKcs synergistically participate in stabilizing replication fork to encounter replication stress in glioma cells. J. Exp. Clin. Cancer Res. 2022, 41, 140. [Google Scholar] [CrossRef]
- Zheng, L.; Dai, H.; Zhou, M.; Li, M.; Singh, P.; Qiu, J.; Tsark, W.; Huang, Q.; Kernstine, K.; Zhang, X.; et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 2007, 13, 812–819. [Google Scholar] [CrossRef]
- Wang, K.; Xie, C.; Chen, D. Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis. Int. J. Mol. Med. 2014, 33, 1268–1274. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.; Zhou, C.; Chen, S. Flap endonuclease 1 (FEN1) as a novel diagnostic and prognostic biomarker for gastric cancer. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Keymeulen, S.; Nelson, R.; Tong, T.R.; Yuan, Y.-C.; Yun, X.; Liu, Z.; Lopez, J.; Raz, D.J.; Kim, J.Y. Overexpression of flap endonuclease 1 correlates with enhanced proliferation and poor prognosis of non-small-cell lung cancer. Am. J. Pathol. 2018, 188, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.G.; Sotiriou, S.K.; Halazonetis, T.D. Draining the FEN1s for cancer therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 21849–21850. [Google Scholar] [CrossRef]
- Guo, E.; Ishii, Y.; Mueller, J.; Srivatsan, A.; Gahman, T.; Putnam, C.D.; Wang, J.Y.J.; Kolodner, R.D. FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proc. Natl. Acad. Sci. USA 2020, 117, 19415–19424. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.A.; Russell, R.; Albarakati, N.; Maloney, D.J.; Dorjsuren, D.; Rueda, O.M.; Moseley, P.; Mohan, V.; Sun, H.; Abbotts, R.; et al. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol. Oncol. 2014, 8, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, P.; Wang, P.; Fang, Z.; Zhu, Y. Identification of flap endonuclease 1 with diagnostic and prognostic value in breast cancer. Front. Oncol. 2021, 11, 603114. [Google Scholar] [CrossRef]
- Xu, L.; Qu, J.-L.; Song, N.; Zhang, L.-Y.; Zeng, X.; Che, X.-F.; Hou, K.-Z.; Shi, S.; Feng, Z.-Y.; Qu, X.-J.; et al. Biological and clinical significance of flap endonuclease-1 in triple-negative breast cancer: Support of metastasis and a poor prognosis. Oncol. Rep. 2020, 44, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ba, S.; Mahajan, D.; Lee, J.Y.; Ye, R.; Shao, F.; Lu, L.; Li, T. Versatile types of DNA-based nanobiosensors for specific detection of cancer biomarker FEN1 in living cells and cell-free systems. Nano Lett. 2018, 18, 7383–7388. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Wu, Y.; Zhou, L.; He, J.; Yang, C.; Zhang, P.; Hu, R.; Luo, C.; Du, J.; Fu, J.; et al. Variants and haplotypes in Flap endonuclease 1 and risk of gallbladder cancer and gallstones: A population-based study in China. Sci. Rep. 2015, 5, 18160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Ni, W.; Zhu, M.; Zhang, X.; Qiang, Y.; Zhang, J.; Ni, Z.; Shen, Y.; Qiu, S.; Song, Q.; et al. Flap endonuclease 1 facilitated hepatocellular carcinoma progression by enhancing USP7/MDM2-mediated P53 inactivation. Int. J. Biol. Sci. 2022, 18, 1022–1038. [Google Scholar] [CrossRef]
- Pu, J.; Wang, J.; Qin, Z.; Wang, A.; Zhang, Y.; Wu, X.; Wu, Y.; Li, W.; Xu, Z.; Lu, Y.; et al. IGF2BP2 promotes liver cancer growth through an m6A-FEN1-dependent mechanism. Front. Oncol. 2020, 10, 578816. [Google Scholar] [CrossRef]
- Al-Kawaz, A.; Miligy, I.M.; Toss, M.S.; Mohammed, O.J.; Green, A.R.; Madhusudan, S.; Rakha, E.A. The prognostic significance of Flap Endonuclease 1 (FEN1) in breast ductal carcinoma in situ. Breast Cancer Res. Treat. 2021, 188, 53–63. [Google Scholar] [CrossRef]
- Li, C.; Zhou, D.; Hong, H.; Yang, S.; Zhang, L.; Li, S.; Hu, P.; Ren, H.; Mei, Z.; Tang, H. TGFβ1- miR-140-5p axis mediated up-regulation of Flap Endonuclease 1 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Aging 2019, 11, 5593–5612. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dai, H.; Wang, Y.; Liang, Y.; Feng, W.; Yuan, Y. Targeting FEN1 suppresses the proliferation of chronic myeloid leukemia cells through regulating alternative end-joining pathways. DNA Cell Biol. 2021, 40, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Gabison, J.; Liu, Y. Trinucleotide repeat deletion via a unique hairpin bypass by DNA polymerase β and alternate flap cleavage by flap endonuclease 1. Nucleic Acids Res. 2013, 41, 1684–1697. [Google Scholar] [CrossRef]
- Wang, J.; Cao, P.; Qi, Y.-Y.; Chen, X.-P.; Ma, L.; Deng, R.-R.; Zhang, L.-L.; Zhao, Y. The relationship between cell apoptosis dysfunction and FEN1 E160D mutation in lupus nephritis patients. Autoimmunity 2017, 50, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Wang, Z.-Z.; Shi, X.-X.; Huang, G.-Y.; Hao, G.-F.; Yang, G.-F. Fragment-based drug design facilitates selective kinase inhibitor discovery. Trends Pharmacol. Sci. 2021, 42, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer. 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Bom, D.; Huck, B.; Gleason, E.; Wang, J.; Silver, D.; Brunden, K.; Boozer, S.; Rundlett, S.; Sherf, B.; et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 277–281. [Google Scholar] [CrossRef]
- He, L.; Luo, L.; Zhu, H.; Yang, H.; Zhang, Y.; Wu, H.; Sun, H.; Jiang, F.; Kathera, C.S.; Liu, L.; et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol. Oncol. 2017, 11, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Flach, K.D.; Periyasamy, M.; Jadhav, A.; Dorjsuren, D.; Siefert, J.C.; Hickey, T.E.; Opdam, M.; Patel, H.; Canisius, S.; Wilson, D.M.; et al. Endonuclease FEN1 coregulates ERα Activity and provides a novel drug interface in tamoxifen-resistant breast cancer. Cancer Res. 2020, 80, 1914–1926. [Google Scholar] [CrossRef] [Green Version]
- McManus, K.J.; Barrett, I.J.; Nouhi, Y.; Hieter, P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc. Natl. Acad. Sci. USA 2009, 106, 3276–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-L.; Wang, J.-P.; Chang, H.; Deng, S.-M.; Du, J.-H.; Wang, X.-X.; Hu, H.-J.; Li, D.-Y.; Xu, X.-B.; Guo, W.-Q.; et al. FEN1 inhibitor increases sensitivity of radiotherapy in cervical cancer cells. Cancer Med. 2019, 8, 7774–7780. [Google Scholar] [CrossRef]
- He, L.; Yang, H.; Zhou, S.; Zhu, H.; Mao, H.; Ma, Z.; Wu, T.; Kumar, A.K.; Kathera, C.; Janardhan, A.; et al. Synergistic antitumor effect of combined paclitaxel with FEN1 inhibitor in cervical cancer cells. DNA Repair. 2018, 63, 1–9. [Google Scholar] [CrossRef]
- Wu, T.; Zhu, H.; Zhang, M.; Sun, Y.; Yang, Y.; Gu, L.; Zhang, J.; Mu, D.; Wu, C.; Hu, Z.; et al. FEN1 inhibitor synergizes with low-dose camptothecin to induce increased cell killing via the mitochondria mediated apoptotic pathway. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Ma, L.; Cao, X.; Wang, H.; Lu, K.; Wang, Y.; Tu, C.; Dai, Y.; Meng, Y.; Li, Y.; Yu, P.; et al. Discovery of myricetin as a potent inhibitor of human flap endonuclease 1, which potentially can be used as sensitizing agent against HT-29 human colon cancer cells. J. Agric. Food Chem. 2019, 67, 1656–1665. [Google Scholar] [CrossRef]
- Ba, S.; Zhang, H.; Lee, J.Y.; Wu, H.; Ye, R.; Huang, D.; Li, T. Investigation of human flap structure-specific endonuclease 1 (FEN1) activity on primer-template models and exploration of a substrate-based FEN1 inhibitor. Bioorg. Med. Chem. 2016, 24, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. small-molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Li, L.; Kumar, A.K.; Hu, Z.; Guo, Z. Small molecule inhibitors targeting key proteins in the DNA damage response for cancer therapy. Curr. Med. Chem. 2021, 28, 963–985. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Hu, Z.; Guo, Z. Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy. Biomolecules 2022, 12, 1007. https://doi.org/10.3390/biom12071007
Yang F, Hu Z, Guo Z. Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy. Biomolecules. 2022; 12(7):1007. https://doi.org/10.3390/biom12071007
Chicago/Turabian StyleYang, Fan, Zhigang Hu, and Zhigang Guo. 2022. "Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy" Biomolecules 12, no. 7: 1007. https://doi.org/10.3390/biom12071007
APA StyleYang, F., Hu, Z., & Guo, Z. (2022). Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy. Biomolecules, 12(7), 1007. https://doi.org/10.3390/biom12071007