
Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics


 and
and
Abstract
:1. Introduction
- (1)
- How robust is the evidence supporting an alteration of the glutamate system and dopamine-glutamate interaction in TRS?
- (2)
- How and to what extent can D-amino acids dysregulation intercept the glutamatergic abnormalities in TRS?
- (3)
- Do preclinical and clinical findings substantiate the D-amino acid strategy in TRS?
- (4)
- What are the putative innovative future trials and targets?
2. Materials and Methods
3. The neurobiology of Treatment-Resistant Schizophrenia and Amino Acid-Related Metabolism
4. Dopamine–Glutamate Interaction and the Role of D-Amino Acids
5. D-Amino Acids as an Innovative Potential Therapeutic Approach to Mitigate NMDAR Hypofunction in Schizophrenia
5.1. NMDAR Co-Agonists
5.2. D-Serine
Clinical Reports of D-Serine Efficacy in Treating Schizophrenia Patients Not Responding to Antipsychotics
5.3. D-Alanine
5.4. D-Aspartate
6. D-Amino Acid Oxidase
7. Other Modulators of the Glycine B Site at NMDAR
7.1. Glycine
7.1.1. Glycine Transport Inhibitors for the Treatment of Schizophrenia
7.1.2. Sarcosine
7.1.3. Non-Sarcosine Derivatives GlyT-1 Inhibitors
7.2. D-Cycloserine
7.3. D-Peptides
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCutcheon, R.A.; Marques, T.R.; Howes, O.D. Schizophrenia-An Overview. JAMA Psychiatry 2020, 77, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Howes, O.D. Treatment-Resistant Schizophrenia: Definition, Predictors, and Therapy Options. J. Clin. Psychiatry 2021, 82, 36608. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McCutcheon, R.; Agid, O.; De Bartolomeis, A.; Van Beveren, N.J.; Birnbaum, M.L.; Bloomfield, M.; Bressan, R.A.; Buchanan, R.W.; Carpenter, W.T.; et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. Am. J. Psychiatry 2017, 174, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.D.; Heaton, R.K.; Carpenter, W.T.; Green, M.F.; Gold, J.M.; Schoenbaum, M. Functional impairment in people with schizophrenia: Focus on employability and eligibility for disability compensation. Schizophr. Res. 2012, 140, 1–8. [Google Scholar] [CrossRef] [Green Version]
- De Bartolomeis, A.; Balletta, R.; Giordano, S.; Buonaguro, E.F.; Latte, G.; Iasevoli, F. Differential cognitive performances between schizophrenic responders and non-responders to antipsychotics: Correlation with course of the illness, psychopathology, attitude to the treatment and antipsychotics doses. Psychiatry Res. 2013, 210, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Avagliano, C.; Altavilla, B.; Barone, A.; D’Ambrosio, L.; Matrone, M.; Francesco, D.N.; Razzino, E.; De Bartolomeis, A. Disease Severity in Treatment Resistant Schizophrenia Patients Is Mainly Affected by Negative Symptoms, Which Mediate the Effects of Cognitive Dysfunctions and Neurological Soft Signs. Front. Psychiatry 2018, 9, 553. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Prinzivalli, E.; Callovini, G.; D’Ambrosio, L.; Altavilla, B.; Avagliano, C.; Iasevoli, F. Treatment resistant schizophrenia and neurological soft signs may converge on the same pathology: Evidence from explanatory analysis on clinical, psychopathological, and cognitive variables. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 356–366. [Google Scholar] [CrossRef]
- Mizuno, K.; Mizuno, E.; Suekane, A.; Shiratsuchi, T. Efficacy of clozapine for long-stay patients with treatment-resistant schizophrenia: 4-year observational study. Neuropsychopharmacol. Rep. 2022, 42, 183–190. [Google Scholar] [CrossRef]
- Correll, C.U.; Martin, A.; Patel, C.; Benson, C.; Goulding, R.; Kern-Sliwa, J.; Joshi, K.; Schiller, E.; Kim, E. Systematic literature review of schizophrenia clinical practice guidelines on acute and maintenance management with antipsychotics. Schizophrenia 2022, 8, 5. [Google Scholar] [CrossRef]
- Gammon, D.; Cheng, C.; Volkovinskaia, A.; Baker, G.; Dursun, S. Clozapine: Why Is It So Uniquely Effective in the Treatment of a Range of Neuropsychiatric Disorders? Biomolecules 2021, 11, 1030. [Google Scholar] [CrossRef]
- Segev, A.; Iqbal, E.; McDonagh, T.A.; Casetta, C.; Oloyede, E.; Piper, S.; Plymen, C.M.; MacCabe, J.H. Clozapine-induced myocarditis: Electronic health register analysis of incidence, timing, clinical markers and diagnostic accuracy. Br. J. Psychiatry 2021, 219, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.M. Clozapine is underutilized. Shanghai Arch. Psychiatry 2012, 24, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Zamanpoor, M. Schizophrenia in a genomic era: A review from the pathogenesis, genetic and environmental etiology to diagnosis and treatment insights. Psychiatr. Genet. 2020, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mäki-Marttunen, T.; Kaufmann, T.; Elvsåshagen, T.; Devor, A.; Djurovic, S.; Westlye, L.T.; Linne, M.-L.; Rietschel, M.; Schubert, D.; Borgwardt, S.; et al. Biophysical Psychiatry—How Computational Neuroscience Can Help to Understand the Complex Mechanisms of Mental Disorders. Front. Psychiatry 2019, 10, 534. [Google Scholar] [CrossRef] [Green Version]
- Wada, M.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Yoshida, K.; Tani, H.; Hirano, Y.; Koike, S.; Sasabayashi, D.; Katayama, H.; et al. Dopaminergic dysfunction and excitatory/inhibitory imbalance in treatment-resistant schizophrenia and novel neuromodulatory treatment. Mol. Psychiatry 2022, 27, 2950–2967. [Google Scholar] [CrossRef]
- Potkin, S.G.; Kane, J.M.; Correll, C.U.; Lindenmayer, J.-P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. Schizophrenia 2020, 6, 1. [Google Scholar] [CrossRef]
- Billard, J.-M. D-Amino acids in brain neurotransmission and synaptic plasticity. Amino Acids 2012, 43, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, H.; Hashimoto, R.; Fujita, Y.; Numata, S.; Yasuda, Y.; Fujimoto, M.; Ohi, K.; Umeda-Yano, S.; Ito, A.; Ohmori, T.; et al. Changes in plasma d-serine, l-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci. Lett. 2014, 582, 93–98. [Google Scholar] [CrossRef]
- Hons, J.; Vasatova, M.; Čermáková, E.; Doubek, P.; Libiger, J. Different serine and glycine metabolism in patients with schizophrenia receiving clozapine. J. Psychiatr. Res. 2012, 46, 811–818. [Google Scholar] [CrossRef]
- Harrison, P.J. D-Amino Acid Oxidase Inhibition: A New Glutamate Twist for Clozapine Augmentation in Schizophrenia? Biol. Psychiatry 2018, 84, 396–398. [Google Scholar] [CrossRef]
- Leppik, L.; Kriisa, K.; Koido, K.; Koch, K.; Kajalaid, K.; Haring, L.; Vasar, E.; Zilmer, M. Profiling of Amino Acids and Their Derivatives Biogenic Amines Before and After Antipsychotic Treatment in First-Episode Psychosis. Front. Psychiatry 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errico, F.; Nuzzo, T.; Carella, M.; Bertolino, A.; Usiello, A. The Emerging Role of Altered d-Aspartate Metabolism in Schizophrenia: New Insights from Preclinical Models and Human Studies. Front. Psychiatry 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errico, F.; Napolitano, F.; Nisticò, R.; Usiello, A. New insights on the role of free d-aspartate in the mammalian brain. Amino Acids 2012, 43, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Molla, G.; Sacchi, S.; Murtas, G. Human D-aspartate Oxidase: A Key Player in D-aspartate Metabolism. Front. Mol. Biosci. 2021, 8, 689719. [Google Scholar] [CrossRef]
- Seetharam, J.C.; Maiti, R.; Mishra, A.; Mishra, B.R. Efficacy and safety of add-on sodium benzoate, a D-amino acid oxidase inhibitor, in treatment of schizophrenia: A systematic review and meta-analysis. Asian J. Psychiatry 2021, 68, 102947. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: A pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412. [Google Scholar] [CrossRef]
- Tsai, G.E.; Yang, P.; Chang, Y.-C.; Chong, M.-Y. D-Alanine Added to Antipsychotics for the Treatment of Schizophrenia. Biol. Psychiatry 2006, 59, 230–234. [Google Scholar] [CrossRef]
- Keller, S.; Punzo, D.; Cuomo, M.; Affinito, O.; Coretti, L.; Sacchi, S.; Florio, E.; Lembo, F.; Carella, M.; Copetti, M.; et al. DNA methylation landscape of the genes regulating D-serine and D-aspartate metabolism in post-mortem brain from controls and subjects with schizophrenia. Sci. Rep. 2018, 8, 10163. [Google Scholar] [CrossRef] [Green Version]
- Abdulbagi, M.; Wang, L.; Siddig, O.; Di, B.; Li, B. D-Amino Acids and D-Amino Acid-Containing Peptides: Potential Disease Biomarkers and Therapeutic Targets? Biomolecules 2021, 11, 1716. [Google Scholar] [CrossRef]
- Taniguchi, K.; Sawamura, H.; Ikeda, Y.; Tsuji, A.; Kitagishi, Y.; Matsuda, S. D-Amino Acids as a Biomarker in Schizophrenia. Diseases 2022, 10, 9. [Google Scholar] [CrossRef]
- Marchi, M.; Galli, G.; Magarini, F.M.; Mattei, G.; Galeazzi, G.M. Sarcosine as an add-on treatment to antipsychotic medication for people with schizophrenia: A systematic review and meta-analysis of randomized controlled trials. Expert Opin. Drug Metab. Toxicol. 2021, 17, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Strzelecki, D.; Podgórski, M.; Kałużyńska, O.; Stefańczyk, L.; Kotlicka-Antczak, M.; Gmitrowicz, A.; Grzelak, P. Adding Sarcosine to Antipsychotic Treatment in Patients with Stable Schizophrenia Changes the Concentrations of Neuronal and Glial Metabolites in the Left Dorsolateral Prefrontal Cortex. Int. J. Mol. Sci. 2015, 16, 24475–24489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Wang, Z.; Deng, D.; Yan, R.; Ju, J.; Zhou, Q. The impact of D-cycloserine and sarcosine on in vivo frontal neural activity in a schizophrenia-like model. BMC Psychiatry 2019, 19, 314. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Su, T.-P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Slifstein, M.; Kegeles, L.S.; Xu, X.; Thompson, J.L.; Urban, N.; Castrillon, J.; Hackett, E.; Bae, S.-A.; Laruelle, M.; Abi-Dargham, A. Striatal and extrastriatal dopamine release measured with PET and [18F] fallypride. Synapse 2010, 64, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef]
- Caravaggio, F.; Porco, N.; Kim, J.; Carmona, E.T.; Brown, E.; Iwata, Y.; Nakajima, S.; Gerretsen, P.; Remington, G.; Graff-Guerrero, A. Measuring Amphetamine-Induced Dopamine Release in Humans: A Comparative Meta-Analysis of [11C]-Raclopride and [11C]-(+)-PHNO Studies. Synapse 2021, 75, e22195. [Google Scholar] [CrossRef]
- Howes, O.D.; Bose, S.K.; Turkheimer, F.; Valli, I.; Egerton, A.; Valmaggia, L.R.; Murray, R.M.; McGuire, P. Dopamine Synthesis Capacity Before Onset of Psychosis: A Prospective [18F]-DOPA PET Imaging Study. Am. J. Psychiatry 2011, 168, 1311–1317. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, A.; Di Maio, A.; Torretta, S.; Garofalo, M.; Giorgelli, V.; Masellis, R.; Nuzzo, T.; Errico, F.; Bertolino, A.; Subramaniam, S.; et al. Abnormal RasGRP1 Expression in the Post-Mortem Brain and Blood Serum of Schizophrenia Patients. Biomolecules 2022, 12, 328. [Google Scholar] [CrossRef]
- Plavén-Sigray, P.; Victorsson, P.I.; Santillo, A.; Matheson, G.J.; Lee, M.; Collste, K.; Fatouros-Bergman, H.; Sellgren, C.M.; Erhardt, S.; Agartz, I.; et al. Thalamic dopamine D2-receptor availability in schizophrenia: A study on antipsychotic-naive patients with first-episode psychosis and a meta-analysis. Mol. Psychiatry 2021, 27, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Demjaha, A.; Murray, R.; McGuire, P.; Kapur, S.; Howes, O. Dopamine Synthesis Capacity in Patients with Treatment-Resistant Schizophrenia. Am. J. Psychiatry 2012, 169, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Howes, O.D.; Veronese, M.; Beck, K.; Seo, S.; Park, J.W.; Lee, J.S.; Lee, Y.-S.; Kwon, J.S. Presynaptic Dopamine Capacity in Patients with Treatment-Resistant Schizophrenia Taking Clozapine: An [18F]DOPA PET Study. Neuropsychopharmacology 2016, 42, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egerton, A.; Murphy, A.; Donocik, J.; Anton, A.; Barker, G.J.; Collier, T.; Deakin, B.; Drake, R.; Eliasson, E.; Emsley, R.; et al. Dopamine and Glutamate in Antipsychotic-Responsive Compared with Antipsychotic-Nonresponsive Psychosis: A Multicenter Positron Emission Tomography and Magnetic Resonance Spectroscopy Study (STRATA). Schizophr. Bull. 2020, 47, 505–516. [Google Scholar] [CrossRef]
- Mouchlianitis, E.; Bloomfield, M.; Law, V.; Beck, K.; Selvaraj, S.; Rasquinha, N.; Waldman, A.D.; Turkheimer, F.; Egerton, A.; Stone, J.; et al. Treatment-Resistant Schizophrenia Patients Show Elevated Anterior Cingulate Cortex Glutamate Compared to Treatment-Responsive. Schizophr. Bull. 2016, 42, 744–752. [Google Scholar] [CrossRef]
- Tarumi, R.; Tsugawa, S.; Noda, Y.; Plitman, E.; Honda, S.; Matsushita, K.; Chavez, S.; Sawada, K.; Wada, M.; Matsui, M.; et al. Levels of glutamatergic neurometabolites in patients with severe treatment-resistant schizophrenia: A proton magnetic resonance spectroscopy study. Neuropsychopharmacology 2019, 45, 632–640. [Google Scholar] [CrossRef]
- Huang, L.-C.; Lin, S.-H.; Tseng, H.-H.; Chen, K.C.; Abdullah, M.; Yang, Y.K. Altered glutamate level and its association with working memory among patients with treatment-resistant schizophrenia (TRS): A proton magnetic resonance spectroscopy study. Psychol. Med. 2022, 1–8. [Google Scholar] [CrossRef]
- Ochi, R.; Plitman, E.; Patel, R.; Tarumi, R.; Iwata, Y.; Tsugawa, S.; Kim, J.; Honda, S.; Noda, Y.; Uchida, H.; et al. Investigating structural subdivisions of the anterior cingulate cortex in schizophrenia, with implications for treatment resistance and glutamatergic levels. J. Psychiatry Neurosci. 2022, 47, E1–E10. [Google Scholar] [CrossRef]
- Matrone, M.; Kotzalidis, G.D.; Romano, A.; Bozzao, A.; Cuomo, I.; Valente, F.; Gabaglio, C.; Lombardozzi, G.; Trovini, G.; Amici, E.; et al. Treatment-resistant schizophrenia: Addressing white matter integrity, intracortical glutamate levels, clinical and cognitive profiles between early- and adult-onset patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 114, 110493. [Google Scholar] [CrossRef]
- Reyes-Madrigal, F.; Guma, E.; León-Ortiz, P.; Gómez-Cruz, G.; Mora-Durán, R.; Graff-Guerrero, A.; Kegeles, L.S.; Chakravarty, M.M.; de la Fuente-Sandoval, C. Striatal glutamate, subcortical structure and clinical response to first-line treatment in first-episode psychosis patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 113, 110473. [Google Scholar] [CrossRef]
- Goldstein, M.E.; Anderson, V.M.; Pillai, A.; Kydd, R.R.; Russell, B.R. Glutamatergic Neurometabolites in Clozapine-Responsive and-Resistant Schizophrenia. Int. J. Neuropsychopharmacol. 2015, 18, pyu117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, J.M.N.; Xin, L. Magnetic Resonance Spectroscopy in Schizophrenia: Evidence for Glutamatergic Dysfunction and Impaired Energy Metabolism. Neurochem. Res. 2018, 44, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, J.S.; Inoue, T.; Hayashi-Takagi, A. Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia. Biomolecules 2020, 10, 1172. [Google Scholar] [CrossRef]
- Mei, Y.-Y.; Wu, D.C.; Zhou, N. Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 2018, 9, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egerton, A.; Grace, A.A.; Stone, J.; Bossong, M.G.; Sand, M.; McGuire, P. Glutamate in schizophrenia: Neurodevelopmental perspectives and drug development. Schizophr. Res. 2020, 223, 59–70. [Google Scholar] [CrossRef]
- Geyer, M.A.; Krebs-Thomson, K.; Braff, D.L.; Swerdlow, N.R. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: A decade in review. Psychopharmacology 2001, 156, 117–154. [Google Scholar] [CrossRef]
- Smith, K.R.; Kopeikina, K.J.; Fawcett-Patel, J.M.; Leaderbrand, K.; Gao, R.; Schürmann, B.; Myczek, K.; Radulovic, J.; Swanson, G.T.; Penzes, P. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 2014, 84, 399–415. [Google Scholar] [CrossRef] [Green Version]
- De Bartolomeis, A.; Avagliano, C.; Vellucci, L.; D’Ambrosio, L.; Manchia, M.; D’Urso, G.; Buonaguro, E.F.; Iasevoli, F. Translating preclinical findings in clinically relevant new antipsychotic targets: Focus on the glutamatergic postsynaptic density. Implications for treatment resistant schizophrenia. Neurosci. Biobehav. Rev. 2019, 107, 795–827. [Google Scholar] [CrossRef]
- Dienel, S.J.; Schoonover, K.E.; Lewis, D.A. Cognitive Dysfunction and Prefrontal Cortical Circuit Alterations in Schizophrenia: Developmental Trajectories. Biol. Psychiatry 2022. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. Schizophrenia 2017, 3, 32. [Google Scholar] [CrossRef]
- MacKay, M.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-Serine: Potential Therapeutic Agent and/or Biomarker in Schizophrenia and Depression? Front. Psychiatry 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abekawa, T.; Ito, K.; Koyama, T. Role of the simultaneous enhancement of NMDA and dopamine D1 receptor-mediated neurotransmission in the effects of clozapine on phencyclidine-induced acute increases in glutamate levels in the rat medial prefrontal cortex. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 374, 177–193. [Google Scholar] [CrossRef]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipina, T.; Labrie, V.; Weiner, I.; Roder, J. Modulators of the glycine site on NMDA receptors, d-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology 2005, 179, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Verrall, L.; Burnet, P.; Betts, J.F.; Harrison, P.J. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol. Psychiatry 2009, 15, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.; Tsukamoto, T. Recent Advances in the Discovery of D-Amino Acid Oxidase Inhibitors and Their Therapeutic Utility in Schizophrenia. Curr. Pharm. Des. 2011, 17, 103–111. [Google Scholar] [CrossRef]
- Seeman, P.; Lee, T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975, 188, 1217–1219. [Google Scholar] [CrossRef]
- Creese, I.; Burt, D.R.; Snyder, S.H. Dopamine Receptor Binding Predicts Clinical and Pharmacological Potencies of Antischizophrenic Drugs. Science 1976, 192, 481–483. [Google Scholar] [CrossRef]
- Olney, J.W.; Newcomer, J.W.; Farber, N. NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 1999, 33, 523–533. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.; Jevtovic-Todorovic, V.; Selke, G.; Melson, A.K.; Hershey, T.; Craft, S.; Olney, J.W. Ketamine-Induced NMDA Receptor Hypofunction as a Model of Memory Impairment and Psychosis. Neuropsychopharmacology 1999, 20, 106–118. [Google Scholar] [CrossRef]
- Farber, N.B.; Wozniak, D.F.; Price, M.T.; Labruyere, J.; Huss, J.; St Peter, H.; Olney, J.W. Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: Potential relevance to schizophrenia? Biol. Psychiatry 1995, 38, 788–796. [Google Scholar] [CrossRef]
- Olney, J.W.; Farber, N.B. NMDA antagonists as neurotherapeutic drugs, psychotogens, neurotoxins, and research tools for studying schizophrenia. Neuropsychopharmacology 1995, 13, 335–345. [Google Scholar] [CrossRef]
- Martinez, Z.A.; Halim, N.D.; Oostwegel, J.L.; Geyer, M.A.; Swerdlow, N.R. Ontogeny of Phencyclidine and Apomorphine-Induced Startle Gating Deficits in Rats. Pharmacol. Biochem. Behav. 2000, 65, 449–457. [Google Scholar] [CrossRef]
- Vollenweider, F.X.; Vontobel, P.; Øye, I.; Hell, D.; Leenders, K.L. Effects of (S)-ketamine on striatal dopamine: A [11C]raclopride PET study of a model psychosis in humans. J. Psychiatr. Res. 2000, 34, 35–43. [Google Scholar] [CrossRef]
- Breier, A.; Adler, C.M.; Weisenfeld, N.; Su, T.-P.; Elman, I.; Picken, L.; Malhotra, A.K.; Pickar, D. Effects of NMDA antagonism on striatal dopamine release in healthy subjects: Application of a novel PET approach. Synapse 1998, 29, 142–147. [Google Scholar] [CrossRef]
- Smith, G.; Schloesser, R.; Brodie, J.; Dewey, S.L.; Logan, J.; Vitkun, S.A.; Simkowitz, P.; Hurley, A.; Cooper, T.; Volkow, N.D.; et al. Glutamate Modulation of Dopamine Measured in Vivo with Positron Emission Tomography (PET) and 11C-Raclopride in Normal Human Subjects. Neuropsychopharmacology 1998, 18, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.M.; Malhotra, A.K.; Elman, I.; Goldberg, T.; Egan, M.; Pickar, D.; Breier, A. Comparison of Ketamine-Induced Thought Disorder in Healthy Volunteers and Thought Disorder in Schizophrenia. Am. J. Psychiatry 1999, 156, 1646–1649. [Google Scholar] [CrossRef]
- Olney, J.W.; Farber, N.B. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [CrossRef]
- Javitt, D.C. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside J. Clin. Psychiatry 1987, 9, 12–35. [Google Scholar]
- Coyle, J.T. The Glutamatergic Dysfunction Hypothesis for Schizophrenia. Harv. Rev. Psychiatry 1996, 3, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Fiore, G.; Iasevoli, F. Dopamine-Glutamate Interaction and Antipsychotics Mechanism of Action: Implication for New Pharmacological Strategies in Psychosis. Curr. Pharm. Des. 2005, 11, 3561–3594. [Google Scholar] [CrossRef]
- Buck, S.A.; Erickson-Oberg, M.Q.; Logan, R.W.; Freyberg, Z. Relevance of interactions between dopamine and glutamate neurotransmission in schizophrenia. Mol. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Parellada, E.; Gassó, P. Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, M.; Carlsson, A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia-implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990, 13, 272–276. [Google Scholar] [CrossRef]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Kegeles, L.S.; Abi-Dargham, A.; Zea-Ponce, Y.; Rodenhiser-Hill, J.; Mann, J.J.; Van Heertum, R.L.; Cooper, T.B.; Carlsson, A.; Laruelle, M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biol. Psychiatry 2000, 48, 627–640. [Google Scholar] [CrossRef]
- Belforte, J.E.; Zsiros, V.; Sklar, E.R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E.M.; Nakazawa, K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010, 13, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Nikolaus, S.; Beu, M.; Wittsack, H.-J.; Müller-Lutz, A.; Antke, C.; Hautzel, H.; Mori, Y.; Mamlins, E.; Antoch, G.; Müller, H.-W. GABAergic and glutamatergic effects on nigrostriatal and mesolimbic dopamine release in the rat. Rev. Neurosci. 2020, 31, 569–588. [Google Scholar] [CrossRef]
- Kim, J.; Horti, A.G.; Mathews, W.B.; Pogorelov, V.; Valentine, H.; Brašić, J.R.; Holt, D.P.; Ravert, H.T.; Dannals, R.F.; Zhou, L.; et al. Quantitative Multi-modal Brain Autoradiography of Glutamatergic, Dopaminergic, Cannabinoid, and Nicotinic Receptors in Mutant Disrupted-In-Schizophrenia-1 (DISC1) Mice. Mol. Imaging Biol. 2014, 17, 355–363. [Google Scholar] [CrossRef]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.D.; Pierce, R.C. Cocaine-induced neuroadaptations in glutamate transmission: Potential therapeutic targets for craving and addiction. Ann. N. Y. Acad. Sci. 2010, 1187, 35–75. [Google Scholar] [CrossRef] [PubMed]
- Greengard, P.; Allen, P.B.; Nairn, A.C. Beyond the Dopamine Receptor: The DARPP-32/Protein Phosphatase-1 Cascade. Neuron 1999, 23, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.C.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The Glutamatergic Aspects of Schizophrenia Molecular Pathophysiology: Role of the Postsynaptic Density, and Implications for Treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.-M.; Wang, Y.-C.; Wu, C.-L.; Hsu, S.-H.; Tsai, H.-Y.; Cheng, M.-C. Multiple Rare Risk Coding Variants in Postsynaptic Density-Related Genes Associated With Schizophrenia Susceptibility. Front. Genet. 2020, 11, 524258. [Google Scholar] [CrossRef]
- Yamauchi, T. Molecular constituents and phosphorylation-dependent regulation of the post-synaptic density. Mass Spectrom. Rev. 2002, 21, 266–286. [Google Scholar] [CrossRef]
- Boeckers, T.M. The postsynaptic density. Cell Tissue Res. 2006, 326, 409–422. [Google Scholar] [CrossRef]
- Gold, M.G. A frontier in the understanding of synaptic plasticity: Solving the structure of the postsynaptic density. BioEssays 2012, 34, 599–608. [Google Scholar] [CrossRef] [Green Version]
- De Bartolomeis, A.; Sarappa, C.; Buonaguro, E.F.; Marmo, F.; Eramo, A.; Tomasetti, C.; Iasevoli, F. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 1–12. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Latte, G.; Tomasetti, C.; Iasevoli, F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol. Neurobiol. 2014, 49, 484–511. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; Ambesi-Impiombato, A.; Muscettola, G.; de Bartolomeis, A. Dopamine receptor subtypes contribution to Homer1a induction: Insights into antipsychotic molecular action. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Dell’Aversano, C.; Iasevoli, F.; de Bartolomeis, A. Homer splice variants modulation within cortico-subcortical regions by dopamine D2 antagonists, a partial agonist, and an indirect agonist: Implication for glutamatergic postsynaptic density in antipsychotics action. Neuroscience 2007, 150, 144–158. [Google Scholar] [CrossRef]
- Barone, A.; Signoriello, S.; Latte, G.; Vellucci, L.; Giordano, G.; Avagliano, C.; Buonaguro, E.F.; Marmo, F.; Tomasetti, C.; Iasevoli, F.; et al. Modulation of glutamatergic functional connectivity by a prototypical antipsychotic: Translational inference from a postsynaptic density immediate-early gene-based network analysis. Behav. Brain Res. 2021, 404, 113160. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Moratalla, R.; Robertson, H.A. Amphetamine and cocaine induce drug-specific activation of the c-fos gene in striosome-matrix compartments and limbic subdivisions of the striatum. Proc. Natl. Acad. Sci. USA 1990, 87, 6912–6916. [Google Scholar] [CrossRef] [Green Version]
- Moratalla, R.; Robertson, H.; Graybiel, A. Dynamic regulation of NGFI-A (zif268, egr1) gene expression in the striatum. J. Neurosci. 1992, 12, 2609–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.V.; Kosofsky, B.E.; Birnbaum, R.; Cohen, B.M.; Hyman, S.E. Differential expression of c-fos and zif268 in rat striatum after haloperidol, clozapine, and amphetamine. Proc. Natl. Acad. Sci. USA 1992, 89, 4270–4274. [Google Scholar] [CrossRef] [Green Version]
- Konradi, C.; Cole, R.L.; Heckers, S.; Hyman, S.E. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J. Neurosci. 1994, 14, 5623–5634. [Google Scholar] [CrossRef] [Green Version]
- Cole, R.L.; Konradi, C.; Douglass, J.; Hyman, S.E. Neuronal adaptation to amphetamine and dopamine: Molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron 1995, 14, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Konradi, C.; Leveque, J.C.; Hyman, S.E. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J. Neurosci. 1996, 16, 4231–4239. [Google Scholar] [CrossRef] [Green Version]
- Keefe, K.A.; Ganguly, A. Effects of NMDA receptor antagonists on D1 dopamine receptor-mediated changes in striatal immediate early gene expression: Evidence for involvement of pharmacologically distinct NMDA receptors? Dev. Neurosci. 1998, 20, 216–228. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Barone, A.; Buonaguro, E.F.; Tomasetti, C.; Vellucci, L.; Iasevoli, F. The Homer1 family of proteins at the crossroad of dopamine-glutamate signaling: An emerging molecular “Lego” in the pathophysiology of psychiatric disorders. A systematic review and translational insight. Neurosci. Biobehav. Rev. 2022, 136, 104596. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzadeh, M.B.; Windham, L.K.; Lake, R.W.; Acker, C.J.; Kalivas, P.W. Cocaine activates Homer1 immediate early gene transcription in the mesocorticolimbic circuit: Differential regulation by dopamine and glutamate signaling. Synapse 2008, 63, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammermeier, P.J.; Worley, P.F. Homer 1a uncouples metabotropic glutamate receptor 5 from postsynaptic effectors. Proc. Natl. Acad. Sci. USA 2007, 104, 6055–6060. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Zeng, W.; Kiselyov, K.; Yuan, J.P.; Dehoff, M.H.; Mikoshiba, K.; Worley, P.F.; Muallem, S. Homer 1 Mediates Store- and Inositol 1,4,5-Trisphosphate Receptor-dependent Translocation and Retrieval of TRPC3 to the Plasma Membrane. J. Biol. Chem. 2006, 281, 32540–32549. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Yang, L.; Tang, Q.; Samdani, S.; Zhang, G.; Wang, J.Q. The Scaffold Protein Homer1b/c Links Metabotropic Glutamate Receptor 5 to Extracellular Signal-Regulated Protein Kinase Cascades in Neurons. J. Neurosci. 2005, 25, 2741–2752. [Google Scholar] [CrossRef]
- Klugmann, M.; Symes, C.W.; Leichtlein, C.B.; Klaussner, B.K.; Dunning, J.; Fong, D.; Young, D.; During, M.J. AAV-mediated hippocampal expression of short and long Homer 1 proteins differentially affect cognition and seizure activity in adult rats. Mol. Cell. Neurosci. 2005, 28, 347–360. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Mizutani, A.; Yuasa, S.; Mikoshiba, K.; Furuichi, T. Glutamate-induced declustering of post-synaptic adaptor protein Cupidin (Homer 2/vesl-2) in cultured cerebellar granule cells. J. Neurochem. 2003, 87, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Kiselyov, K.; Shin, D.M.; Chen, J.; Shcheynikov, N.; Kang, S.H.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Muallem, S.; et al. Homer Binds TRPC Family Channels and Is Required for Gating of TRPC1 by IP3 Receptors. Cell 2003, 114, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Hennou, S.; Kato, A.; Schneider, E.M.; Lundstrom, K.; Gahwiler, B.H.; Inokuchi, K.; Gerber, U.; Ehrengruber, M.U. Homer-1a/Vesl-1S enhances hippocampal synaptic transmission. Eur. J. Neurosci. 2003, 18, 811–819. [Google Scholar] [CrossRef]
- Sala, C.; Futai, K.; Yamamoto, K.; Worley, P.F.; Hayashi, Y.; Sheng, M. Inhibition of Dendritic Spine Morphogenesis and Synaptic Transmission by Activity-Inducible Protein Homer1a. J. Neurosci. 2003, 23, 6327–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ango, F.; Robbe, D.; Tu, J.C.; Xiaob, B.; Worley, P.F.; Pin, J.-P.; Bockaert, J.; Fagnia, L. Homer-Dependent Cell Surface Expression of Metabotropic Glutamate Receptor Type 5 in Neurons. Mol. Cell. Neurosci. 2002, 20, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Sergé, A.; Fourgeaud, L.; Hémar, A.; Choquet, D. Receptor Activation and Homer Differentially Control the Lateral Mobility of Metabotropic Glutamate Receptor 5 in the Neuronal Membrane. J. Neurosci. 2002, 22, 3910–3920. [Google Scholar] [CrossRef] [Green Version]
- Sala, C.; Piëch, V.; Wilson, N.R.; Passafaro, M.; Liu, G.; Sheng, M. Regulation of Dendritic Spine Morphology and Synaptic Function by Shank and Homer. Neuron 2001, 31, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Ango, F.; Prézeau, L.; Muller, T.; Tu, J.C.; Xiao, B.; Worley, P.F.; Pin, J.-P.; Bockaert, J.; Fagni, L. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 2001, 411, 962–965. [Google Scholar] [CrossRef]
- Foa, L.; Rajan, I.; Haas, K.; Wu, G.-Y.; Brakeman, P.; Worley, P.; Cline, H. The scaffold protein, Homer1b/c, regulates axon pathfinding in the central nervous system in vivo. Nat. Neurosci. 2001, 4, 499–506. [Google Scholar] [CrossRef]
- Kammermeier, P.J.; Xiao, B.; Tu, J.C.; Worley, P.F.; Ikeda, S. Homer Proteins Regulate Coupling of Group I Metabotropic Glutamate Receptors to N-Type Calcium and M-Type Potassium Channels. J. Neurosci. 2000, 20, 7238–7245. [Google Scholar] [CrossRef]
- Ango, F.; Pin, J.-P.; Tu, J.C.; Xiao, B.; Worley, P.F.; Bockaert, J.; Fagni, L. Dendritic and Axonal Targeting of Type 5 Metabotropic Glutamate Receptor Is Regulated by Homer1 Proteins and Neuronal Excitation. J. Neurosci. 2000, 20, 8710–8716. [Google Scholar] [CrossRef] [Green Version]
- Ciruela, F.; Soloviev, M.M.; Chan, W.-Y.; McIlhinney, R. Homer-1c/Vesl-1L Modulates the Cell Surface Targeting of Metabotropic Glutamate Receptor Type 1α: Evidence for an Anchoring Function. Mol. Cell. Neurosci. 2000, 15, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, S.; Tachibana, T.; Imanaka, T.; Nishida, W.; Sobue, K. Involvement of unique leucine-zipper motif of PSD-Zip45 (Homer 1c/vesl-1L) in group 1 metabotropic glutamate receptor clustering. Proc. Natl. Acad. Sci. USA 1999, 96, 13801–13806. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Mizutani, A.; Bito, H.; Fujisawa, K.; Narumiya, S.; Mikoshiba, K.; Furuichi, T. Cupidin, an Isoform of Homer/Vesl, Interacts with the Actin Cytoskeleton and Activated Rho Family Small GTPases and Is Expressed in Developing Mouse Cerebellar Granule Cells. J. Neurosci. 1999, 19, 8389–8400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, K.W.; Tu, J.C.; Petralia, R.S.; Xiao, B.; Wenthold, R.J.; Worley, P.F. Homer 1b Regulates the Trafficking of Group I Metabotropic Glutamate Receptors. J. Biol. Chem. 1999, 274, 25953–25957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciruela, F.; Soloviev, M.M.; McIlhinney, R.A. Co-expression of metabotropic glutamate receptor type 1alpha with homer-1a/Vesl-1S increases the cell surface expression of the receptor. Biochem. J. 1999, 341 Pt 3, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.C.; Xiao, B.; Yuan, J.P.; Lanahan, A.A.; Leoffert, K.; Li, M.; Linden, D.J.; Worley, P.F. Homer Binds a Novel Proline-Rich Motif and Links Group 1 Metabotropic Glutamate Receptors with IP3 Receptors. Neuron 1998, 21, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Tu, J.C.; Petralia, R.S.; Yuan, J.P.; Doan, A.; Breder, C.D.; Ruggiero, A.; Lanahan, A.A.; Wenthold, R.J.; Worley, P.F. Homer Regulates the Association of Group 1 Metabotropic Glutamate Receptors with Multivalent Complexes of Homer-Related, Synaptic Proteins. Neuron 1998, 21, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Lominac, K.; Oleson, E.B.; Pava, M.; Klugmann, M.; Schwarz, M.K.; Seeburg, P.H.; During, M.J.; Worley, P.F.; Kalivas, P.W.; Szumlinski, K.K. Distinct Roles for Different Homer1 Isoforms in Behaviors and Associated Prefrontal Cortex Function. J. Neurosci. 2005, 25, 11586–11594. [Google Scholar] [CrossRef]
- Tappe, A.; Kuner, R. Regulation of motor performance and striatal function by synaptic scaffolding proteins of the Homer1 family. Proc. Natl. Acad. Sci. USA 2006, 103, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, T.X.; Hallett, P.J.; Watanabe, M.; Grant, S.G.; Isacson, O.; Yao, W.D. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J. Neurosci. 2009, 29, 2948–2960. [Google Scholar] [CrossRef]
- Zhang, J.; Saur, T.; Duke, A.N.; Grant, S.G.N.; Platt, D.M.; Rowlett, J.K.; Isacson, O.; Yao, W.-D. Motor Impairments, Striatal Degeneration, and Altered Dopamine-Glutamate Interplay in Mice Lacking PSD-95. J. Neurogenetics 2014, 28, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Destreel, G.; Seutin, V.; Engel, D. Subsaturation of the N-methyl-D-aspartate receptor glycine site allows the regulation of bursting activity in juvenile rat nigral dopamine neurons. Eur. J. Neurosci. 2019, 50, 3454–3471. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [Green Version]
- De Bartolomeis, A.; Errico, F.; Aceto, G.; Tomasetti, C.; Usiello, A.; Iasevoli, F. D-aspartate dysregulation in Ddo(−/−) mice modulates phencyclidine-induced gene expression changes of postsynaptic density molecules in cortex and striatum. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 62, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Dallérac, G.; Li, X.; Lecouflet, P.; Morisot, N.; Sacchi, S.; Asselot, R.; Pham, T.H.; Potier, B.; Watson, D.J.G.; Schmidt, S.; et al. Dopaminergic neuromodulation of prefrontal cortex activity requires the NMDA receptor coagonist d-serine. Proc. Natl. Acad. Sci. USA 2021, 118, e2023750118. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Puhl, M.D.; Anderson, T.; Balu, D.T.; Coyle, J.T. Serine Racemase Expression by Striatal Neurons. Cell. Mol. Neurobiol. 2020, 42, 279–289. [Google Scholar] [CrossRef]
- Collingridge, G.; Abraham, W. Glutamate receptors and synaptic plasticity: The impact of Evans and Watkins. Neuropharmacology 2021, 206, 108922. [Google Scholar] [CrossRef] [PubMed]
- Kleckner, N.W.; Dingledine, R. Requirement for Glycine in Activation of NMDA-Receptors Expressed in Xenopus Oocytes. Science 1988, 241, 835–837. [Google Scholar] [CrossRef]
- Zhang, B.; Xiong, F.; Ma, Y.; Li, B.; Mao, Y.; Zhou, Z.; Yu, H.; Li, J.; Li, C.; Fu, J.; et al. Chronic phencyclidine treatment impairs spatial working memory in rhesus monkeys. Psychopharmacology 2019, 236, 2223–2232. [Google Scholar] [CrossRef]
- Aniline, O.; Pitts, F.N. Phencyclidine (PCP): A Review and Perspectives. CRC Crit. Rev. Toxicol. 1982, 10, 145–177. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Javitt, D.C. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: The final common pathway on the road to schizophrenia? Brain Res. Bull. 2010, 83, 108–121. [Google Scholar] [CrossRef] [Green Version]
- De Bartolomeis, A.; Manchia, M.; Marmo, F.; Vellucci, L.; Iasevoli, F.; Barone, A. Glycine Signaling in the Framework of Dopamine-Glutamate Interaction and Postsynaptic Density. Implications for Treatment-Resistant Schizophrenia. Front. Psychiatry 2020, 11, 369. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Dumas, T.C. Developmental regulation of cognitive abilities: Modified composition of a molecular switch turns on associative learning. Prog. Neurobiol. 2005, 76, 189–211. [Google Scholar] [CrossRef] [PubMed]
- Lussier, M.; Sanz-Clemente, A.; Roche, K.W. Dynamic Regulation of N-Methyl-d-aspartate (NMDA) and α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptors by Posttranslational Modifications. J. Biol. Chem. 2015, 290, 28596–28603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafra, F.; Ibáñez, I.; Bartolomé-Martín, D.; Piniella, D.; Arribas-Blázquez, M.; Giménez, C. Glycine Transporters and Its Coupling with NMDA Receptors. Adv. Neurobiol. 2017, 16, 55–83. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lládo, J.; Calderó, J.; Ribera, J.; Tarabal, O.; Oppenheim, R.W.; Esquerda, J.E. Opposing Effects of Excitatory Amino Acids on Chick Embryo Spinal Cord Motoneurons: Excitotoxic Degeneration or Prevention of Programmed Cell Death. J. Neurosci. 1999, 19, 10803–10812. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. NMDA receptors: Recent insights and clinical correlations. Neurology 2011, 76, 1750–1757. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Wada, A.; Yoshida, K.; Miyoshi, Y.; Sayano, T.; Esaki, K.; Kinoshita, M.O.; Tomonaga, S.; Azuma, N.; Watanabe, M.; et al. Brain-specific Phgdh Deletion Reveals a Pivotal Role for l-Serine Biosynthesis in Controlling the Level of d-Serine, an N-methyl-d-aspartate Receptor Co-agonist, in Adult Brain. J. Biol. Chem. 2010, 285, 41380–41390. [Google Scholar] [CrossRef] [Green Version]
- Henneberger, C.; Bard, L.; Rusakov, D.A. D-Serine: A key to synaptic plasticity? Int. J. Biochem. Cell Biol. 2012, 44, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, E.R.; Gustafson, E.C.; Miller, R.F. Glycine transport accounts for the differential role of glycine vs. d-serine at NMDA receptor coagonist sites in the salamander retina. Eur. J. Neurosci. 2010, 31, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, E.C.; Stevens, E.R.; Wolosker, H.; Miller, R.F. Endogenous d-Serine Contributes to NMDA-Receptor–Mediated Light-Evoked Responses in the Vertebrate Retina. J. Neurophysiol. 2007, 98, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ge, W.; Chen, Y.; Zhang, Z.; Shen, W.; Wu, C.; Poo, M.; Duan, S. Contribution of astrocytes to hippocampal long-term potentiation through release of d-serine. Proc. Natl. Acad. Sci. USA 2003, 100, 15194–15199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mothet, J.-P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.-A.; Sekiguchi, M.; Hashimoto, A.; Tomita, U.; Nishikawa, T.; Wada, K. Functional Comparison of d-Serine and Glycine in Rodents: The Effect on Cloned NMDA Receptors and the Extracellular Concentration. J. Neurochem. 2002, 65, 454–458. [Google Scholar] [CrossRef]
- Mothet, J.P.; Rouaud, E.; Sinet, P.-M.; Potier, B.; Jouvenceau, A.; Dutar, P.; Videau, C.; Epelbaum, J.; Billard, J.-M. A critical role for the glial-derived neuromodulator d-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell 2006, 5, 267–274. [Google Scholar] [CrossRef]
- Panatier, A.; Theodosis, D.T.; Mothet, J.-P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H. Glia-Derived d-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef]
- Berger, A.J.; Dieudonné, S.; Ascher, P. Glycine Uptake Governs Glycine Site Occupancy at NMDA Receptors of Excitatory Synapses. J. Neurophysiol. 1998, 80, 3336–3340. [Google Scholar] [CrossRef] [Green Version]
- Schell, M.J.; Brady, R.O., Jr.; Molliver, M.E.; Snyder, S.H. D-Serine as a Neuromodulator: Regional and Developmental Localizations in Rat Brain Glia Resemble NMDA Receptors. J. Neurosci. 1997, 17, 1604–1615. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, A.; Oka, T.; Nishikawa, T. Extracellular concentration of endogenous free d-serine in the rat brain as revealed by in vivo microdialysis. Neuroscience 1995, 66, 635–643. [Google Scholar] [CrossRef]
- Basu, A.C.; Tsai, G.E.; Ma, C.-L.; Ehmsen, J.T.; Mustafa, A.K.; Han, L.; Jiang, Z.I.; Benneyworth, M.A.; Froimowitz, M.P.; Lange, N.; et al. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 2008, 14, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jami, S.A.; Cameron, S.; Wong, J.M.; Daly, E.R.; McAllister, A.K.; Gray, J.A. Increased excitation-inhibition balance and loss of GABAergic synapses in the serine racemase knockout model of NMDA receptor hypofunction. J. Neurophysiol. 2021, 126, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Labrie, V.; Fukumura, R.; Rastogi, A.; Fick, L.J.; Wang, W.; Boutros, P.C.; Kennedy, J.L.; Semeralul, M.O.; Lee, F.H.; Baker, G.B.; et al. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 2009, 18, 3227–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matveeva, T.M.; Pisansky, M.T.; Young, A.; Miller, R.F.; Gewirtz, J.C. Sociality deficits in serine racemase knockout mice. Brain Behav. 2019, 9, e01383. [Google Scholar] [CrossRef] [Green Version]
- Labrie, V.; Lipina, T.; Roder, J.C. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology 2008, 200, 217–230. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial d-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. J. Cereb. Blood Flow Metab. 2011, 165, 1543–1555. [Google Scholar] [CrossRef] [Green Version]
- Martina, M.; Krasteniakov, N.V.; Bergeron, R. D-Serine differently modulates NMDA receptor function in rat CA1 hippocampal pyramidal cells and interneurons. J. Physiol. 2003, 548 Pt 2, 411–423. [Google Scholar] [CrossRef]
- Chapman, D.E.; Keefe, K.A.; Wilcox, K.S. Evidence for Functionally Distinct Synaptic NMDA Receptors in Ventromedial Versus Dorsolateral Striatum. J. Neurophysiol. 2003, 89, 69–80. [Google Scholar] [CrossRef]
- Panizzutti, R.; Rausch, M.; Zurbrügg, S.; Baumann, D.; Beckmann, N.; Rudin, M. The pharmacological stimulation of NMDA receptors via co-agonist site: An fMRI study in the rat brain. Neurosci. Lett. 2005, 380, 111–115. [Google Scholar] [CrossRef]
- Burnet, P.; Hutchinson, L.; Von Hesling, M.; Gilbert, E.-J.; Brandon, N.; Rutter, A.; Hutson, P.; Harrison, P. Expression of D-serine and glycine transporters in the prefrontal cortex and cerebellum in schizophrenia. Schizophr. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef]
- Hashimoto, A.; Yoshikawa, M. Effect of aminooxyacetic acid on extracellular level of d-serine in rat striatum: An in vivo microdialysis study. Eur. J. Pharmacol. 2005, 525, 91–93. [Google Scholar] [CrossRef]
- Bendikov, I.; Nadri, C.; Amar, S.; Panizzutti, R.; De Miranda, J.; Wolosker, H.; Agam, G. A CSF and postmortem brain study of d-serine metabolic parameters in schizophrenia. Schizophr. Res. 2007, 90, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Ohnishi, T.; Hashimoto, K.; Ohba, H.; Iwayama-Shigeno, Y.; Toyoshima, M.; Okuno, A.; Takao, H.; Toyota, T.; Minabe, Y.; et al. Identification of Multiple Serine Racemase (SRR) mRNA Isoforms and Genetic Analyses of SRR and DAO in Schizophrenia and d-Serine Levels. Biol. Psychiatry 2005, 57, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Hons, J.; Zirko, R.; Ulrychova, M.; Cermakova, E.; Libiger, J. D-serine serum levels in patients with schizophrenia: Relation to psychopathology and comparison to healthy subjects. Neuro Endocrinol. Lett. 2008, 29, 485–492. [Google Scholar]
- Fuchs, S.A.; De Barse, M.M.; Scheepers, F.E.; Cahn, W.; Dorland, L.; Velden, M.G.D.S.-V.D.; Klomp, L.W.; Berger, R.; Kahn, R.S.; de Koning, T.J. Cerebrospinal fluid d-serine and glycine concentrations are unaltered and unaffected by olanzapine therapy in male schizophrenic patients. Eur. Neuropsychopharmacol. 2008, 18, 333–338. [Google Scholar] [CrossRef]
- Ono, K.; Shishido, Y.; Park, H.K.; Kawazoe, T.; Iwana, S.; Chung, S.P.; El-Magd, R.M.A.; Yorita, K.; Okano, M.; Watanabe, T.; et al. Potential pathophysiological role of d-amino acid oxidase in schizophrenia: Immunohistochemical and in situ hybridization study of the expression in human and rat brain. J. Neural Transm. 2009, 116, 1335–1347. [Google Scholar] [CrossRef]
- Ozeki, Y.; Sekine, M.; Fujii, K.; Watanabe, T.; Okayasu, H.; Takano, Y.; Shinozaki, T.; Aoki, A.; Akiyama, K.; Homma, H.; et al. Phosphoserine phosphatase activity is elevated and correlates negatively with plasma d-serine concentration in patients with schizophrenia. Psychiatry Res. 2016, 237, 344–350. [Google Scholar] [CrossRef]
- De Rosa, A.; Fontana, A.; Nuzzo, T.; Garofalo, M.; Di Maio, A.; Punzo, D.; Copetti, M.; Bertolino, A.; Errico, F.; Rampino, A.; et al. Machine Learning algorithm unveils glutamatergic alterations in the post-mortem schizophrenia brain. Schizophrenia 2022, 8, 8. [Google Scholar] [CrossRef]
- Burnet, P.W.J.; Eastwood, S.L.; Bristow, G.C.; Godlewska, B.R.; Sikka, P.; Walker, M.; Harrison, P.J. D-Amino acid oxidase activity and expression are increased in schizophrenia. Mol. Psychiatry 2008, 13, 658–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habl, G.; Zink, M.; Petroianu, G.; Bauer, M.; Schneider-Axmann, T.; von Wilmsdorff, M.; Falkai, P.; Henn, F.A.; Schmitt, A. Increased d-amino acid oxidase expression in the bilateral hippocampal CA4 of schizophrenic patients: A post-mortem study. J. Neural Transm. 2009, 116, 1657–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, C.; Freitas, M.E.; Vargas-Lopes, C.; Wolosker, H.; Panizzutti, R. Increased brain d-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr. Res. 2008, 101, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Verrall, L.; Walker, M.; Rawlings, N.; Benzel, I.; Kew, J.N.C.; Harrison, P.J.; Burnet, P.W.J. D-Amino acid oxidase and serine racemase in human brain: Normal distribution and altered expression in schizophrenia. Eur. J. Neurosci. 2007, 26, 1657–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, M.J.; Molliver, M.E.; Snyder, S.H. D-serine, an endogenous synaptic modulator: Localization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. USA 1995, 92, 3948–3952. [Google Scholar] [CrossRef] [Green Version]
- Foltyn, V.N.; Bendikov, I.; De Miranda, J.; Panizzutti, R.; Dumin, E.; Shleper, M.; Li, P.; Toney, M.D.; Kartvelishvily, E.; Wolosker, H. Serine racemase modulates intracellular D-serine levels through an α,β-elimination activity. J. Biol. Chem. 2005, 280, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Hikida, T.; Mustafa, A.K.; Maeda, K.; Fujii, K.; Barrow, R.K.; Saleh, M.; Huganir, R.L.; Snyder, S.H.; Hashimoto, K.; Sawa, A. Modulation of d-Serine Levels in Brains of Mice Lacking PICK. Biol. Psychiatry 2008, 63, 997–1000. [Google Scholar] [CrossRef] [Green Version]
- Fujii, K.; Maeda, K.; Hikida, T.; Mustafa, A.K.; Balkissoon, R.; Xia, J.; Yamada, T.; Ozeki, Y.; Kawahara, R.; Okawa, M.; et al. Serine racemase binds to PICK1: Potential relevance to schizophrenia. Mol. Psychiatry 2005, 11, 150–157. [Google Scholar] [CrossRef]
- Wang, X.; He, G.; Gu, N.; Yang, J.; Tang, J.; Chen, Q.; Liu, X.; Shen, Y.; Qian, X.; Lin, W.; et al. Association of G72/G30 with schizophrenia in the Chinese population. Biochem. Biophys Res. Commun. 2004, 319, 1281–1286. [Google Scholar] [CrossRef]
- Schumacher, J.; Jamra, R.A.; Freudenberg, J.; Becker, T.; Ohlraun, S.; Otte, A.C.J.; Tullius, M.; Kovalenko, S.; Bogaert, A.V.D.; Maier, W.; et al. Examination of G72 and D-amino-acid oxidase as genetic risk factors for schizophrenia and bipolar affective disorder. Mol. Psychiatry 2004, 9, 203–207. [Google Scholar] [CrossRef]
- Addington, A.M.; Gornick, M.; Sporn, A.L.; Gogtay, N.; Greenstein, D.; Lenane, M.; Gochman, P.; Baker, N.; Balkissoon, R.; Vakkalanka, R.K.; et al. Polymorphisms in the 13q33.2 gene G72/G30 are associated with childhood-onset schizophrenia and psychosis not otherwise specified. Biol. Psychiatry 2004, 55, 976–980. [Google Scholar] [CrossRef]
- Hattori, E.; Liu, C.; Badner, J.A.; Bonner, T.I.; Christian, S.L.; Maheshwari, M.; Detera-Wadleigh, S.D.; Gibbs, R.A.; Gershon, E.S. Polymorphisms at the G72/G30 Gene Locus, on 13q33, Are Associated with Bipolar Disorder in Two Independent Pedigree Series*. Am. J. Hum. Genet. 2003, 72, 1131–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumakov, I.; Blumenfeld, M.; Guerassimenko, O.; Cavarec, L.; Palicio, M.; Abderrahim, H.; Bougueleret, L.; Barry, C.; Tanaka, H.; La Rosa, P.; et al. Genetic and physiological data implicating the new human gene G72 and the gene for d-amino acid oxidase in schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 13675–13680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.S.; Akula, N.; Detera-Wadleigh, S.D.; Schulze, T.G.; Thomas, J.; Potash, J.B.; DePaulo, J.R.; McInnis, M.G.; Cox, N.J.; McMahon, F.J. Findings in an independent sample support an association between bipolar affective disorder and the G72/G30 locus on chromosome 13q. Mol. Psychiatry 2004, 9, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balu, D.T.; Coyle, J.T. Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 2011, 35, 848–870. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.M.; Abazyan, S.; Abazyan, B.; Nomura, J.; Yang, C.; Seshadri, S.J.; Sawa, A.; Snyder, S.H.; Pletnikov, M.V. Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol. Psychiatry 2012, 18, 557–567. [Google Scholar] [CrossRef]
- Svane, K.C.; Asis, E.-K.; Omelchenko, A.; Kunnath, A.J.; Brzustowicz, L.M.; Silverstein, S.M.; Firestein, B.L. D-Serine administration affects nitric oxide synthase 1 adaptor protein and DISC1 expression in sex-specific manner. Mol. Cell. Neurosci. 2018, 89, 20–32. [Google Scholar] [CrossRef]
- Brzustowicz, L.M.; Simone, J.; Mohseni, P.; Hayter, J.E.; Hodgkinson, K.A.; Chow, E.W.; Bassett, A.S. Linkage Disequilibrium Mapping of Schizophrenia Susceptibility to the CAPON Region of Chromosome 1q. Am. J. Hum. Genet. 2004, 74, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Carrel, D.; Du, Y.; Komlos, D.; Hadzimichalis, N.M.; Kwon, M.; Wang, B.; Brzustowicz, L.M.; Firestein, B.L. NOS1AP Regulates Dendrite Patterning of Hippocampal Neurons through a Carboxypeptidase E-Mediated Pathway. J. Neurosci. 2009, 29, 8248–8258. [Google Scholar] [CrossRef]
- Li, L.-L.; De Mera, R.M.M.-F.; Chen, J.; Ba, W.; Kasri, N.N.; Zhang, M.; Courtney, M.J.; De Mera, R.M.M.F. Unexpected Heterodivalent Recruitment of NOS1AP to nNOS Reveals Multiple Sites for Pharmacological Intervention in Neuronal Disease Models. J. Neurosci. 2015, 35, 7349–7364. [Google Scholar] [CrossRef] [Green Version]
- Hadzimichalis, N.M.; Previtera, M.L.; Moreau, M.P.; Li, B.; Lee, G.H.; Dulencin, A.M.; Matteson, P.G.; Buyske, S.; Millonig, J.H.; Brzustowicz, L.M.; et al. NOS1AP protein levels are altered in BA46 and cerebellum of patients with schizophrenia. Schizophr. Res. 2010, 124, 248–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastwood, S.L. Does the CAPON gene confer susceptibility to schizophrenia? PLoS Med. 2005, 2, e348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, H.; Iyo, M.; Hashimoto, K. Neonatal Disruption of Serine Racemase Causes Schizophrenia-Like Behavioral Abnormalities in Adulthood: Clinical Rescue by D-Serine. PLoS ONE 2013, 8, e62438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, S.-I.; Ishii, S.; Umino, A.; Shimazu, D.; Yamamoto, N.; Nishikawa, T. Effects of psychotomimetic and antipsychotic agents on neocortical and striatal concentrations of various amino acids in the rat. J. Neurochem. 2004, 90, 1378–1388. [Google Scholar] [CrossRef]
- Panizzutti, R.; Fisher, M.; Garrett, C.; Man, W.H.; Sena, W.; Madeira, C.; Vinogradov, S. Association between increased serum d-serine and cognitive gains induced by intensive cognitive training in schizophrenia. Schizophr. Res. 2018, 207, 63–69. [Google Scholar] [CrossRef]
- Ohnuma, T.; Sakai, Y.; Maeshima, H.; Hatano, T.; Hanzawa, R.; Abe, S.; Kida, S.; Shibata, N.; Suzuki, T.; Arai, H. Changes in plasma glycine, l-serine, and d-serine levels in patients with schizophrenia as their clinical symptoms improve: Results from the Juntendo University Schizophrenia Projects (JUSP). Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1905–1912. [Google Scholar] [CrossRef]
- Hons, J.; Zirko, R.; Vasatova, M.; Doubek, P.; Klimova, B.; Masopust, J.; Valis, M.; Kuca, K. Impairment of Executive Functions Associated With Lower D-Serine Serum Levels in Patients With Schizophrenia. Front. Psychiatry 2021, 12, 514579. [Google Scholar] [CrossRef]
- Vardigan, J.D.; Huszar, S.L.; McNaughton, C.H.; Hutson, P.H.; Uslaner, J.M. MK-801 produces a deficit in sucrose preference that is reversed by clozapine, D-serine, and the metabotropic glutamate 5 receptor positive allosteric modulator CDPPB: Relevance to negative symptoms associated with schizophrenia? Pharmacol. Biochem. Behav. 2010, 95, 223–229. [Google Scholar] [CrossRef]
- Karasawa, J.-I.; Hashimoto, K.; Chaki, S. D-Serine and a glycine transporter inhibitor improve MK-801-induced cognitive deficits in a novel object recognition test in rats. Behav. Brain Res. 2008, 186, 78–83. [Google Scholar] [CrossRef]
- Bado, P.; Madeira, C.; Vargas-Lopes, C.; Moulin, T.C.; Wasilewska-Sampaio, A.P.; Maretti, L.; de Oliveira, R.V.; Amaral, O.B.; Panizzutti, R. Effects of low-dose d-serine on recognition and working memory in mice. Psychopharmacology 2011, 218, 461–470. [Google Scholar] [CrossRef]
- Dsouza, D.C.; Radhakrishnan, R.; Perry, E.; Bhakta, S.G.; Singh, N.M.; Yadav, R.; Abi-Saab, D.; Pittman, B.; Chaturvedi, S.K.; Sharma, M.P.; et al. Feasibility, Safety, and Efficacy of the Combination of D-Serine and Computerized Cognitive Retraining in Schizophrenia: An International Collaborative Pilot Study. Neuropsychopharmacology 2012, 38, 492–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Epstein, M.; Lee, M.; Lehrfeld, N.; Nolan, K.A.; Shope, C.; Petkova, E.; Silipo, G.; Javitt, D.C. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: Correlation with symptoms. Schizophr. Res. 2018, 191, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Chang, Y.C.; Liu, Y.C.; Chiu, C.C.; Tsai, G.E. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: A randomized, double-blind, placebo-controlled study. Arch. Gen. Psychiatry 2005, 62, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, H.Y.; Lin, C.H.; Huang, Y.J.; Liao, C.H.; Chang, Y.C.; Tsai, G.E. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int. J. Neuropsychopharmacol. 2010, 13, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Tsai, G.; Yang, P.; Chung, L.-C.; Lange, N.; Coyle, J.T. D-serine added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 1998, 44, 1081–1089. [Google Scholar] [CrossRef]
- Tsai, G.E.; Yang, P.; Chung, L.C.; Tsai, I.C.; Tsai, C.W.; Coyle, J.T. D-serine added to clozapine for the treatment of schizophrenia. Am. J. Psychiatry 1999, 156, 1822–1825. [Google Scholar] [CrossRef]
- Weiser, M.; Heresco-Levy, U.; Davidson, M.; Javitt, D.C.; Werbeloff, N.; Gershon, A.A.; Abramovich, Y.; Amital, D.; Doron, A.; Konas, S.; et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J. Clin. Psychiatry 2012, 73, e728–e734. [Google Scholar] [CrossRef]
- Goh, K.K.; Wu, T.H.; Chen, C.H.; Lu, M.L. Efficacy of N-methyl-D-aspartate receptor modulator augmentation in schizophrenia: A meta-analysis of randomised, placebo-controlled trials. J. Psychopharmacol. 2021, 35, 236–252. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Malhotra, A.K.; Cornblatt, B.; Silipo, G.; Balla, A.; Suckow, R.F.; D’Souza, C.; Saksa, J.; Woods, S.W.; Javitt, D.C. High dose D-serine in the treatment of schizophrenia. Schizophr. Res. 2010, 121, 125–130. [Google Scholar] [CrossRef] [Green Version]
- D-Serine AudRem: R33 Phase. Available online: https://clinicaltrials.gov/ct2/show/NCT05046353?term=NCT05046353&draw=2&rank=1 (accessed on 14 June 2022).
- Chung, S.P.; Sogabe, K.; Park, H.K.; Song, Y.; Ono, K.; El-Magd, R.A.; Shishido, Y.; Yorita, K.; Sakai, T.; Fukui, K. Potential cytotoxic effect of hydroxypyruvate produced from D-serine by astroglial D-amino acid oxidase. J. Biochem. 2010, 148, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Shishido, Y.; Ichise-Shishido, S.; Kawazoe, T.; Ono, K.; Iwana, S.; Tomita, Y.; Yorita, K.; Sakai, T.; Fukui, K. Potential Role for Astroglial d-Amino Acid Oxidase in Extracellular d-Serine Metabolism and Cytotoxicity. J. Biochem. 2006, 139, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Carone, F.A.; Nakamura, S.; Goldman, B. Urinary loss of glucose, phosphate, and protein by diffusion into proximal straight tubules injured by D-serine and maleic acid. Lab. Investig. 1985, 52, 605–610. [Google Scholar]
- Adage, T.; Trillat, A.-C.; Quattropani, A.; Perrin, D.; Cavarec, L.; Shaw, J.; Guerassimenko, O.; Giachetti, C.; Gréco, B.; Chumakov, I.; et al. In vitro and in vivo pharmacological profile of AS057278, a selective d-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur. Neuropsychopharmacol. 2008, 18, 200–214. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fujita, Y.; Horio, M.; Kunitachi, S.; Iyo, M.; Ferraris, D.; Tsukamoto, T. Co-Administration of a D-Amino Acid Oxidase Inhibitor Potentiates the Efficacy of D-Serine in Attenuating Prepulse Inhibition Deficits After Administration of Dizocilpine. Biol. Psychiatry 2009, 65, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Uslaner, J.M.; Yao, L.; Mullins, C.M.; Surles, N.O.; Huszar, S.L.; McNaughton, C.H.; Pascarella, D.M.; Kandebo, M.; Hinchliffe, R.M.; et al. The behavioral and neurochemical effects of a novel D-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and D-serine. J. Pharmacol. Exp. Ther. 2009, 328, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duplantier, A.J.; Becker, S.L.; Bohanon, M.J.; Borzilleri, K.A.; Chrunyk, B.A.; Downs, J.T.; Hu, L.-Y.; El-Kattan, A.; James, L.C.; Liu, S.; et al. Discovery, SAR, and Pharmacokinetics of a Novel 3-Hydroxyquinolin-2(1H)-one Series of Potent d-Amino Acid Oxidase (DAAO) Inhibitors. J. Med. Chem. 2009, 52, 3576–3585. [Google Scholar] [CrossRef]
- The Effect of D-Serine as Add-on Therapy in Recent-Onset Psychosis. Available online: https://clinicaltrials.gov/ct2/show/NCT04140773?term=NCT04140773&draw=2&rank=1 (accessed on 14 June 2022).
- Fisher, G.H.; D’Aniello, A.; Vetere, A.; Padula, L.; Cusano, G.P.; Man, E.H. Free D-aspartate and D-alanine in normal and Alzheimer brain. Brain Res. Bull. 1991, 26, 983–985. [Google Scholar] [CrossRef]
- Nagata, Y.; Kubota, K. A trial to determine D-amino acids in tissue proteins of mice. Amino Acids 1993, 4, 121–125. [Google Scholar] [CrossRef]
- Nagata, Y.; Konno, R.; Niwa, A. Amino acid levels in d-alanine-administered mutant mice lacking d-amino acid oxidase. Metabolism 1994, 43, 1153–1157. [Google Scholar] [CrossRef]
- Morikawa, A.; Hamase, K.; Zaitsu, K. Determination of d-alanine in the rat central nervous system and periphery using column-switching high-performance liquid chromatography. Anal. Biochem. 2003, 312, 66–72. [Google Scholar] [CrossRef]
- Sasabe, J.; Suzuki, M. Emerging Role of D-Amino Acid Metabolism in the Innate Defense. Front. Microbiol. 2018, 9, 933. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, Y.; Katane, M.; Miyamoto, T.; Sekine, M.; Sakai-Kato, K.; Homma, H. d-Serine and d-Alanine Regulate Adaptive Foraging Behavior in Caenorhabditis elegans via the NMDA Receptor. J. Neurosci. 2020, 40, 7531–7544. [Google Scholar] [CrossRef] [PubMed]
- McBain, C.J.; Kleckner, N.W.; Wyrick, S.; Dingledine, R. Structural requirements for activation of the glycine coagonist site of N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Mol. Pharmacol. 1989, 36, 556–565. [Google Scholar] [PubMed]
- Chairoungdua, A.; Kanai, Y.; Matsuo, H.; Inatomi, J.; Kim, D.K.; Endou, H. Identification and Characterization of a Novel Member of the Heterodimeric Amino Acid Transporter Family Presumed to be Associated with an Unknown Heavy Chain. J. Biol. Chem. 2001, 276, 49390–49399. [Google Scholar] [CrossRef] [Green Version]
- Rojas, C.; Alt, J.; Ator, N.A.; Wilmoth, H.; Rais, R.; Hin, N.; DeVivo, M.; Popiolek, M.; Tsukamoto, T.; Slusher, B.S. Oral administration of D-alanine in monkeys robustly increases plasma and cerebrospinal fluid levels but experimental D-amino acid oxidase inhibitors had minimal effect. J. Psychopharmacol. 2016, 30, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Popiolek, M.; Tierney, B.; Steyn, S.J.; De Vivo, M. Lack of Effect of Sodium Benzoate at Reported Clinical Therapeutic Concentration on d-Alanine Metabolism in Dogs. ACS Chem. Neurosci. 2018, 9, 2832–2837. [Google Scholar] [CrossRef]
- Tanii, Y.; Nishikawa, T.; Hashimoto, A.; Takahashi, K. Stereoselective antagonism by enantiomers of alanine and serine of phencyclidine-induced hyperactivity, stereotypy and ataxia in the rat. J. Pharmacol. Exp. Ther. 1994, 269, 1040–1048. [Google Scholar]
- Hashimoto, A.; Nishikawa, T.; Oka, T.; Takahashi, K. D-alanine inhibits methamphetamine-induced hyperactivity in rats. Eur. J. Pharmacol. 1991, 202, 105–107. [Google Scholar]
- Umino, A.; Takahashi, K.; Nishikawa, T. Characterization of the phencyclidine-induced increase in prefrontal cortical dopamine metabolism in the rat. J. Cereb. Blood Flow Metab. 1998, 124, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Hatano, T.; Ohnuma, T.; Sakai, Y.; Shibata, N.; Maeshima, H.; Hanzawa, R.; Suzuki, T.; Arai, H. Plasma alanine levels increase in patients with schizophrenia as their clinical symptoms improve—Results from the Juntendo University Schizophrenia Projects (JUSP). Psychiatry Res. 2010, 177, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine racemase: A glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-d-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [Green Version]
- D’aniello, A.; Di Fiore, M.M.; Fisher, G.H.; Milone, A.; Seleni, A.; D’Aniello, S.; Perna, A.; Ingrosso, D. Occurrence of D-aspartic acid and N-methyl-D-aspartic acid in rat neuroendocrine tissues and their role in the modulation of luteinizing hormone and growth hormone release. FASEB J. 2000, 14, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Kumashiro, S.; Nishikawa, T.; Oka, T.; Takahashi, K.; Mito, T.; Takashima, S.; Doi, N.; Mizutani, Y.; Yamazaki, T.; et al. Embryonic Development and Postnatal Changes in Free d-Aspartate and d-Serine in the Human Prefrontal Cortex. J. Neurochem. 1993, 61, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Topo, E.; Soricelli, A.; Di Maio, A.; D’Aniello, E.; Di Fiore, M.M.; D’Aniello, A. Evidence for the involvement of d-aspartic acid in learning and memory of rat. Amino Acids 2009, 38, 1561–1569. [Google Scholar] [CrossRef]
- Spinelli, P.; Brown, E.R.; Ferrandino, G.; Branno, M.; Montarolo, P.G.; D’Aniello, E.; Rastogi, R.K.; D’Aniello, B.; Baccari, G.C.; Fisher, G.; et al. D-aspartic acid in the nervous system ofAplysia limacina: Possible role in neurotransmission. J. Cell. Physiol. 2005, 206, 672–681. [Google Scholar] [CrossRef]
- D’Aniello, S.; Somorjai, I.; Garcia-Fernàndez, J.; Topo, E.; D’Aniello, A. D-Aspartic acid is a novel endogenous neurotransmitter. FASEB J. 2010, 25, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuka, S.; Hayashi, M.; Muroyama, A.; Otsuka, M.; Kozaki, S.; Yamada, H.; Moriyama, Y. d-Aspartate Is Stored in Secretory Granules and Released through a Ca2+-dependent Pathway in a Subset of Rat Pheochromocytoma PC12 Cells. J. Biol. Chem. 2001, 276, 26589–26596. [Google Scholar] [CrossRef] [Green Version]
- Palacín, M.; Estevez, R.; Bertran, J.; Zorzano, A. Molecular Biology of Mammalian Plasma Membrane Amino Acid Transporters. Physiol. Rev. 1998, 78, 969–1054. [Google Scholar] [CrossRef]
- Hashimoto, A.; Oka, T.; Nishikawa, T. Anatomical Distribution and Postnatal Changes in Endogenous Free D-Aspartate and D-Serine in Rat Brain and Periphery. Eur. J. Neurosci. 1995, 7, 1657–1663. [Google Scholar] [CrossRef]
- Schell, M.J.; Cooper, O.B.; Snyder, S.H. D-aspartate localizations imply neuronal and neuroendocrine roles. Proc. Natl. Acad. Sci. USA 1997, 94, 2013–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errico, F.; Cuomo, M.; Canu, N.; Caputo, V.; Usiello, A. New insights on the influence of free d-aspartate metabolism in the mammalian brain during prenatal and postnatal life. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140471. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, A.; Mastrostefano, F.; Di Maio, A.; Nuzzo, T.; Saitoh, Y.; Katane, M.; Isidori, A.M.; Caputo, V.; Marotta, P.; Falco, G.; et al. Prenatal expression of d-aspartate oxidase causes early cerebral d-aspartate depletion and influences brain morphology and cognitive functions at adulthood. Amino Acids 2020, 52, 597–617. [Google Scholar] [CrossRef]
- Olverman, H.; Jones, A.; Mewett, K.; Watkins, J. Structure/activity relations of N-methyl-d-aspartate receptor ligands as studied by their inhibition of [3H]d2-amino-5-phosphonopentanoic acid binding in rat brain membranes. Neuroscience 1988, 26, 17–31. [Google Scholar] [CrossRef]
- Krashia, P.; Ledonne, A.; Nobili, A.; Cordella, A.; Errico, F.; Usiello, A.; D’Amelio, M.; Mercuri, N.B.; Guatteo, E.; Carunchio, I. Persistent elevation of D-Aspartate enhances NMDA receptor-mediated responses in mouse substantia nigra pars compacta dopamine neurons. Neuropharmacology 2016, 103, 69–78. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P. D-Aspartate oxidase, a peroxisomal enzyme in liver of rat and man. Biochim. Biophys. Acta (BBA) Gen. Subj. 1991, 1073, 203–208. [Google Scholar] [CrossRef]
- Katane, M.; Kawata, T.; Nakayama, K.; Saitoh, Y.; Kaneko, Y.; Matsuda, S.; Saitoh, Y.; Miyamoto, T.; Sekine, M.; Homma, H. Characterization of the Enzymatic and Structural Properties of Human D-Aspartate Oxidase and Comparison with Those of the Rat and Mouse Enzymes. Biol. Pharm. Bull. 2015, 38, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Negri, A.; Ceciliani, F.; Tedeschi, G.; Simonic, T.; Ronchi, S. The primary structure of the flavoprotein D-aspartate oxidase from beef kidney. J. Biol. Chem. 1992, 267, 11865–11871. [Google Scholar] [CrossRef]
- Molla, G.; Sacchi, S.; Bernasconi, M.; Pilone, M.S.; Fukui, K.; Polegioni, L. Characterization of human D-amino acid oxidase. FEBS Lett. 2006, 580, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Molla, G.; Chaves-Sanjuan, A.; Savinelli, A.; Nardini, M.; Pollegioni, L. Structure and kinetic properties of human d-aspartate oxidase, the enzyme-controlling d-aspartate levels in brain. FASEB J. 2020, 34, 1182–1197. [Google Scholar] [CrossRef] [Green Version]
- Errico, F.; Nisticò, R.; Palma, G.; Federici, M.; Affuso, A.; Brilli, E.; Topo, E.; Centonze, D.; Bernardi, G.; Bozzi, Y.; et al. Increased levels of d-aspartate in the hippocampus enhance LTP but do not facilitate cognitive flexibility. Mol. Cell. Neurosci. 2008, 37, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Nisticò, R.; Di Giorgio, A.; Squillace, M.; Vitucci, D.; Galbusera, A.; Piccinin, S.; Mango, D.; Fazio, L.; Middei, S.; et al. Free D-aspartate regulates neuronal dendritic morphology, synaptic plasticity, gray matter volume and brain activity in mammals. Transl. Psychiatry 2014, 4, e417. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, G.; Pietracupa, S.; Di Menna, L.; Pescatori, L.; Usiello, A.; Battaglia, G.; Nicoletti, F.; Bruno, V. D-Aspartate activates mGlu receptors coupled to polyphosphoinositide hydrolysis in neonate rat brain slices. Neurosci. Lett. 2010, 478, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Luongo, L.; Squillace, M.; Paolone, G.; Mango, D.; Piccinin, S.; Zianni, E.; Imperatore, R.; Iannotta, M.; Longo, F.; et al. d-Aspartate oxidase influences glutamatergic system homeostasis in mammalian brain. Neurobiol. Aging 2015, 36, 1890–1902. [Google Scholar] [CrossRef]
- Gong, X.-Q.; Frandsen, A.; Lu, W.-Y.; Wan, Y.; Zabek, R.L.; Pickering, D.S.; Bai, D. D-Aspartate and NMDA, but not L-aspartate, block AMPA receptors in rat hippocampal neurons. J. Cereb. Blood Flow Metab. 2005, 145, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Errico, F.; Napolitano, F.; Squillace, M.; Vitucci, D.; Blasi, G.; de Bartolomeis, A.; Bertolino, A.; D’Aniello, A.; Usiello, A. Decreased levels of d-aspartate and NMDA in the prefrontal cortex and striatum of patients with schizophrenia. J. Psychiatr. Res. 2013, 47, 1432–1437. [Google Scholar] [CrossRef]
- Errico, F.; Mothet, J.-P.; Usiello, A. D-Aspartate: An endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J. Pharm. Biomed. Anal. 2015, 116, 7–17. [Google Scholar] [CrossRef]
- Nuzzo, T.; Sacchi, S.; Errico, F.; Keller, S.; Palumbo, O.; Florio, E.; Punzo, D.; Napolitano, F.; Copetti, M.; Carella, M.; et al. Decreased free d-aspartate levels are linked to enhanced d-aspartate oxidase activity in the dorsolateral prefrontal cortex of schizophrenia patients. Schizophrenia 2017, 3, 16. [Google Scholar] [CrossRef]
- Errico, F.; Rossi, S.; Napolitano, F.; Catuogno, V.; Topo, E.; Fisone, G.; D’Aniello, A.; Centonze, D.; Usiello, A. D-Aspartate Prevents Corticostriatal Long-Term Depression and Attenuates Schizophrenia-Like Symptoms Induced by Amphetamine and MK. J. Neurosci. 2008, 28, 10404–10414. [Google Scholar] [CrossRef] [Green Version]
- Sacchi, S.; De Novellis, V.; Paolone, G.; Nuzzo, T.; Iannotta, M.; Belardo, C.; Squillace, M.; Bolognesi, P.; Rosini, E.; Motta, Z.; et al. Olanzapine, but not clozapine, increases glutamate release in the prefrontal cortex of freely moving mice by inhibiting D-aspartate oxidase activity. Sci. Rep. 2017, 7, 46288. [Google Scholar] [CrossRef] [Green Version]
- Errico, F.; Nisticò, R.; Napolitano, F.; Oliva, A.B.; Romano, R.; Barbieri, F.; Florio, T.; Russo, C.; Mercuri, N.B.; Usiello, A. Persistent increase of d-aspartate in d-aspartate oxidase mutant mice induces a precocious hippocampal age-dependent synaptic plasticity and spatial memory decay. Neurobiol. Aging 2009, 32, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Melville, G.W.; Siegler, J.C.; Marshall, P.W.M. The effects of d-aspartic acid supplementation in resistance-trained men over a three month training period: A randomised controlled trial. PLoS ONE 2017, 12, e0182630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melville, G.W.; Siegler, J.C.; Marshall, P.W. Three and six grams supplementation of d-aspartic acid in resistance trained men. J. Int. Soc. Sports Nutr. 2015, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollegioni, L.; Diederichs, K.; Molla, G.; Umhau, S.; Welte, W.; Ghisla, S.; Pilone, M.S. Yeast d-Amino Acid Oxidase: Structural Basis of its Catalytic Properties. J. Mol. Biol. 2002, 324, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Pollegioni, L.; Piubelli, L.; Sacchi, S.; Pilone, M.S.; Molla, G. Physiological functions of D-amino acid oxidases: From yeast to humans. Cell. Mol. Life Sci. 2007, 64, 1373–1394. [Google Scholar] [CrossRef] [PubMed]
- Konno, R.; Sasaki, M.; Asakura, S.; Fukui, K.; Enami, J.; Niwa, A. D-Amino-acid oxidase is not present in the mouse liver. Biochim. Biophys. Acta (BBA) Gen. Subj. 1997, 1335, 173–181. [Google Scholar] [CrossRef]
- Arnold, G.; Liscum, L.; Holtzman, E. Ultrastructural localization of D-amino acid oxidase in microperoxisomes of the rat nervous system. J. Histochem. Cytochem. 1979, 27, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Urai, Y.; Jinnouchi, O.; Kwak, K.T.; Suzue, A.; Nagahiro, S.; Fukui, K. Gene expression of D-amino acid oxidase in cultured rat astrocytes: Regional and cell type specific expression. Neurosci. Lett. 2002, 324, 101–104. [Google Scholar] [CrossRef]
- Horiike, K.; Tojo, H.; Arai, R.; Yamano, T.; Nozaki, M.; Maeda, T. Localization of D-amino acid oxidase in Bergmann glial cells and astrocytes of rat cerebellum. Brain Res. Bull. 1987, 19, 587–596. [Google Scholar] [CrossRef]
- Jagannath, V.; Marinova, Z.; Monoranu, C.-M.; Walitza, S.; Grünblatt, E. Expression of D-Amino Acid Oxidase (DAO/DAAO) and D-Amino Acid Oxidase Activator (DAOA/G72) during Development and Aging in the Human Post-mortem Brain. Front. Neuroanat. 2017, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, R.; Lim, K.S.; Cheng, A.; Garrick, T.; Kapoor, V. Preliminary evidence for a link between schizophrenia and NMDA-glycine site receptor ligand metabolic enzymes, d-amino acid oxidase (DAAO) and kynurenine aminotransferase-1 (KAT-1). Brain Res 2006, 1106, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, D.; Hasan, S.; Tam, S.K.; Engle, S.J.; Brandon, N.J.; Sharp, T.; Foster, R.G.; Harrison, P.J.; Bannerman, D.M.; Peirson, S.N. d-amino acid oxidase knockout (Dao(−/−)) mice show enhanced short-term memory performance and heightened anxiety, but no sleep or circadian rhythm disruption. Eur. J. Neurosci. 2015, 41, 1167–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrie, V.; Wang, W.; Barger, S.W.; Baker, G.B.; Roder, J.C. Genetic loss of D-amino acid oxidase activity reverses schizophrenia-like phenotypes in mice. Genes Brain Behav. 2010, 9, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Chen, X.; Wang, Z.J.; Xu, Q.; Appel-Cresswell, S.; McKeown, M.J. A Genetically Informed, Group fMRI Connectivity Modeling Approach: Application to Schizophrenia. IEEE Trans. Biomed. Eng. 2013, 61, 946–956. [Google Scholar] [CrossRef]
- Papagni, S.A.; Mechelli, A.; Prata, D.P.; Kambeitz, J.; Fu, C.H.; Picchioni, M.; Walshe, M.; Toulopoulou, T.; Bramon, E.; Murray, R.M.; et al. Differential effects of DAAO on regional activation and functional connectivity in schizophrenia, bipolar disorder and controls. NeuroImage 2011, 56, 2283–2291. [Google Scholar] [CrossRef]
- Shishikura, M.; Hakariya, H.; Iwasa, S.; Yoshio, T.; Ichiba, H.; Yorita, K.; Fukui, K.; Fukushima, T. Evaluation of human D-amino acid oxidase inhibition by anti-psychotic drugs in vitro. Biosci. Trends 2014, 8, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, S.; Tabara, H.; Song, Z.; Nakabayashi, M.; Yokoyama, Y.; Fukushima, T. Inhibition of D-Amino Acid Oxidase Activity by Antipsychotic Drugs Evaluated by a Fluorometric Assay Using D-Kynurenine as Substrate. Yakugaku Zasshi 2011, 131, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Abou El-Magd, R.M.; Park, H.K.; Kawazoe, T.; Iwana, S.; Ono, K.; Chung, S.P.; Miyano, M.; Yorita, K.; Sakai, T.; Fukui, K. The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia. J. Psychopharmacol. 2010, 24, 1055–1067. [Google Scholar] [CrossRef]
- Karoum, F.; Freed, W.J.; Chuang, L.-W.; Cannon-Spoor, E.; Wyatt, R.J.; Costa, E. D-DOPA and l-DOPA similarly elevate brain dopamine and produce turning behavior in rats. Brain Res. 1988, 440, 190–194. [Google Scholar] [CrossRef]
- Shindo, H.; Maeda, T. Studies on the Metabolism of D-and L-Isomers of 3, 4-Dihydroxyphenylalanine (DOPA). VI. Metabolism of D-DOPA in Rat Kidney. Chem. Pharm. Bull. 1974, 22, 1721–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Zhou, X.-J.; Konno, R.; Wang, Y.-X. D-dopa is unidirectionally converted to l-dopa by d-amino acid oxidase, followed by dopa transaminase. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.F.; Schweimer, J.; Burnham, K.E.; Burnet, P.; Sharp, T.; Harrison, P.J. D-amino acid oxidase is expressed in the ventral tegmental area and modulates cortical dopamine. Front. Synaptic Neurosci. 2014, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweimer, J.V.; Coullon, G.S.; Betts, J.F.; Burnet, P.W.; Engle, S.J.; Brandon, N.J.; Harrison, P.J.; Sharp, T. Increased burst-firing of ventral tegmental area dopaminergic neurons in D-amino acid oxidase knockout mice in vivo. Eur. J. Neurosci. 2014, 40, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Yang, H.-T.; Chiu, C.-C.; Lane, H.-Y. Blood levels of D-amino acid oxidase vs. D-amino acids in reflecting cognitive aging. Sci. Rep. 2017, 7, 14849. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Chou, W.-H.; Tsou, H.-H.; Fang, C.-P.; Liu, T.-H.; Tsao, H.-H.; Hsu, W.-C.; Weng, Y.-C.; Wang, Y.; Liu, Y.-L. A Post-hoc Study of D-Amino Acid Oxidase in Blood as an Indicator of Post-stroke Dementia. Front. Neurol. 2019, 10, 402. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Liang, S.H.; Xu, Y.; Moore, A.; Ran, C. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci. Rep. 2016, 6, 35613. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, D.; Duvall, B.; Ko, Y.-S.; Thomas, A.G.; Rojas, C.; Majer, P.; Hashimoto, K.; Tsukamoto, T. Synthesis and Biological Evaluation of d-Amino Acid Oxidase Inhibitors. J. Med. Chem. 2008, 51, 3357–3359. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, C.H.; Chang, Y.C.; Huang, Y.J.; Chen, P.W.; Yang, H.T.; Lane, H.Y. Sodium Benzoate, a D-Amino Acid Oxidase Inhibitor, Added to Clozapine for the Treatment of Schizophrenia: A Randomized, Double-Blind, Placebo-Controlled Trial. Biol. Psychiatry 2018, 84, 422–432. [Google Scholar] [CrossRef]
- Lin, C.Y.; Liang, S.Y.; Chang, Y.C.; Ting, S.Y.; Kao, C.L.; Wu, Y.H.; Tsai, G.E.; Lane, H.Y. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: A randomised, double-blind, placebo-controlled trial. World J. Biol. Psychiatry 2017, 18, 357–368. [Google Scholar] [CrossRef]
- Matsuura, A.; Fujita, Y.; Iyo, M.; Hashimoto, K. Effects of sodium benzoate on pre-pulse inhibition deficits and hyperlocomotion in mice after administration of phencyclidine. Acta Neuropsychiatr. 2015, 27, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Jensen, K.; Houang, E.; McRobb, F.M.; Bhat, S.; Svensson, M.; Bochevarov, A.; Day, T.; Dahlgren, M.K.; Bell, J.A.; et al. Discovery of a Novel Class of d-Amino Acid Oxidase Inhibitors Using the Schrödinger Computational Platform. J. Med. Chem. 2022, 65, 6775–6802. [Google Scholar] [CrossRef] [PubMed]
- A Study to Evaluate Efficacy, Safety, Tolerability, and Pharmacokinetics of 3 Dose Levels of TAK-831 in Adjunctive Treatment of Adult Participants with Negative Symptoms of Schizophrenia. Available online: https://clinicaltrials.gov/ct2/show/NCT03382639?id=NCT03382639&draw=2&rank=1&load=cart (accessed on 14 June 2022).
- CLINICAL TRIALS SND12. Available online: http://syneurxtrials.com/Trials/Refractory-Schizophrenia (accessed on 14 June 2022).
- Lane, H.Y.; Lin, C.H.; Green, M.F.; Hellemann, G.; Huang, C.C.; Chen, P.W.; Tun, R.; Chang, Y.C.; Tsai, G.E. Add-on treatment of benzoate for schizophrenia: A randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry 2013, 70, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G.; Baker, A.; Lim, C.C.W.; Foley, S.; Dark, F.; Gordon, A.; Ward, D.; Richardson, D.; Bruxner, G.; Beckmann, K.M.; et al. Effect of Sodium Benzoate vs. Placebo among Individuals with Early Psychosis: A Randomized Clinical Trial. JAMA Netw. Open. 2020, 3, e2024335. [Google Scholar] [CrossRef]
- Naas, E.; Zilles, K.; Gnahn, H.; Betz, H.; Becker, C.-M.; Schröder, H. Glycine receptor immunoreactivity in rat and human cerebral cortex. Brain Res. 1991, 561, 139–146. [Google Scholar] [CrossRef]
- Gielen, M.C.; Thomas, P.; Smart, T.G. The desensitization gate of inhibitory Cys-loop receptors. Nat. Commun. 2015, 6, 6829. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.J.; Vandenberg, R.J. Glycine Transporters and Receptors as Targets for Analgesics. Biomolecules 2021, 11, 1676. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Corradini, L.; Nicholson, J.; Smith, M. Assessment of the Anti-Allodynic and Anti-Hyperalgesic Efficacy of a Glycine Transporter 2 Inhibitor Relative to Pregabalin, Duloxetine and Indomethacin in a Rat Model of Cisplatin-Induced Peripheral Neuropathy. Biomolecules 2021, 11, 940. [Google Scholar] [CrossRef]
- Cioffi, C. Inhibition of Glycine Re-Uptake: A Potential Approach for Treating Pain by Augmenting Glycine-Mediated Spinal Neurotransmission and Blunting Central Nociceptive Signaling. Biomolecules 2021, 11, 864. [Google Scholar] [CrossRef]
- Zeilhofer, H.U.; Werynska, K.; Gingras, J.; Yévenes, G.E. Glycine Receptors in Spinal Nociceptive Control—An Update. Biomolecules 2021, 11, 846. [Google Scholar] [CrossRef]
- Barsch, L.; Werdehausen, R.; Leffler, A.; Eulenburg, V. Modulation of Glycinergic Neurotransmission may Contribute to the Analgesic Effects of Propacetamol. Biomolecules 2021, 11, 493. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Ermilov, M.; Lichtenberg, P.; Bar, G.; Javitt, D.C. High-dose glycine added to olanzapine and risperidone for the treatment of schizophrenia. Biol. Psychiatry 2004, 55, 165–171. [Google Scholar] [CrossRef]
- Pinard, E.; Borroni, E.; Koerner, A.; Umbricht, D.; Alberati, D. Glycine Transporter Type I (GlyT1) Inhibitor, Bitopertin: A Journey from Lab to Patient. CHIMIA 2018, 72, 477–484. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Kaufman, M.J.; Cohen, B.M.; Jensen, J.E.; Coyle, J.T.; Du, F.; Öngür, D. In Vivo Brain Glycine and Glutamate Concentrations in Patients with First-Episode Psychosis Measured by Echo Time–Averaged Proton Magnetic Resonance Spectroscopy at 4T. Biol. Psychiatry 2018, 83, 484–491. [Google Scholar] [CrossRef]
- Cioffi, C.L.; Guzzo, P.R. Inhibitors of Glycine Transporter-1: Potential Therapeutics for the Treatment of CNS Disorders. Curr. Top. Med. Chem. 2016, 16, 3404–3437. [Google Scholar] [CrossRef]
- Tsai, G.; Ralph-Williams, R.J.; Martina, M.; Bergeron, R.; Berger-Sweeney, J.; Dunham, K.S.; Jiang, Z.; Caine, S.B.; Coyle, J.T. Gene knockout of glycine transporter 1: Characterization of the behavioral phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Hons, J.; Zirko, R.; Ulrychova, M.; Cermakova, E.; Doubek, P.; Libiger, J. Glycine serum level in schizophrenia: Relation to negative symptoms. Psychiatry Res. 2010, 176, 103–108. [Google Scholar] [CrossRef]
- Neeman, G.; Blanaru, M.; Bloch, B.; Kremer, I.; Ermilov, M.; Javitt, D.C.; Heresco-Levy, U. Relation of Plasma Glycine, Serine, and Homocysteine Levels to Schizophrenia Symptoms and Medication Type. Am. J. Psychiatry 2005, 162, 1738–1740. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Bar, G.; Levin, R.; Ermilov, M.; Ebstein, R.P.; Javitt, D.C. High glycine levels are associated with prepulse inhibition deficits in chronic schizophrenia patients. Schizophr. Res. 2007, 91, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Serrita, J.; Ralevski, E.; Yoon, G.; Petrakis, I. A Pilot Randomized, Placebo-Controlled Trial of Glycine for Treatment of Schizophrenia and Alcohol Dependence. J. Dual Diagn. 2019, 15, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Jin, Y.; Bunney, B.G.; Costa, J.; Gulasekaram, B. Effect of Clozapine and Adjunctive High-Dose Glycine in Treatment-Resistant Schizophrenia. Am. J. Psychiatry 1999, 156, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Zylberman, I.; Zukin, S.R.; Heresco-Levy, U.; Lindenmayer, J.P. Amelioration of negative symptoms in schizophrenia by glycine. Am. J. Psychiatry 1994, 151, 1234–1236. [Google Scholar] [CrossRef]
- Javitt, D.C.; Silipo, G.; Cienfuegos, A.; Shelley, A.-M.; Bark, N.; Park, M.; Lindenmayer, J.-P.; Suckow, R.; Zukin, S.R. Adjunctive high-dose glycine in the treatment of schizophrenia. Int. J. Neuropsychopharmacol. 2001, 4, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Horowitz, A.; Kelly, D. Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. Br. J. Psychiatry 1996, 169, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Silipo, G.; Lichtenstein, M. Efficacy of High-Dose Glycine in the Treatment of Enduring Negative Symptoms of Schizophrenia. Arch. Gen. Psychiatry 1999, 56, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, L.-M.; Leung, S.; Michie, P.T.; Green, A.; Nathan, P.J.; Fitzgerald, P.; Johnston, P.; Solowij, N.; Kulkarni, J.; Croft, R.J. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr. Res. 2017, 191, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Evins, A.E. Placebo-Controlled Trial of Glycine Added to Clozapine in Schizophrenia. Am. J. Psychiatry 2000, 157, 826–828. [Google Scholar] [CrossRef]
- Diaz, P.; Bhaskara, S.; Dursun, S.M.; Deakin, B. Double-blind, Placebo-Controlled, Crossover Trial of Clozapine Plus Glycine in Refractory Schizophrenia Negative Results. J. Clin. Psychopharmacol. 2005, 25, 277–278. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Javitt, D.C.; Marder, S.R.; Schooler, N.R.; Gold, J.M.; McMahon, R.P.; Heresco-Levy, U.; Carpenter, W.T. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): The Efficacy of Glutamatergic Agents for Negative Symptoms and Cognitive Impairments. Am. J. Psychiatry 2007, 164, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.; Khaled, E.; Sramek, J.; Bunney, W.; Potkin, S.G. An Open Trial of Glycine as an Adjunct to Neuroleptics in Chronic Treatment-Refractory Schizophrenics. J. Clin. Psychopharmacol. 1990, 10, 71–72. [Google Scholar] [CrossRef]
- Rosse, R.B.; Theut, S.K.; Banay-Schwartz, M.; Leighton, M.; Scarcella, E.; Cohen, C.G.; Deutsch, S.I. Glycine adjuvant therapy to conventional neuroleptic treatment in schizophrenia: An open-label, pilot study. Clin. Neuropharmacol. 1989, 12, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Leiderman, E.; Zylberman, I.; Zukin, S.; Cooper, T.B.; Javitt, D.C. Preliminary investigation of high-dose oral glycine on serum levels and negative symptoms in schizophrenia: An open-label trial. Biol. Psychiatry 1996, 39, 213–215. [Google Scholar] [CrossRef]
- Lane, H.-Y.; Huang, C.-L.; Wu, P.-L.; Liu, Y.-C.; Chang, Y.-C.; Lin, P.-Y.; Chen, P.-W.; Tsai, G. Glycine Transporter I Inhibitor, N-methylglycine (Sarcosine), Added to Clozapine for the Treatment of Schizophrenia. Biol. Psychiatry 2006, 60, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Dravid, S.M.; Burger, P.B.; Prakash, A.; Geballe, M.T.; Yadav, R.; Le, P.; Vellano, K.; Snyder, J.P.; Traynelis, S.F. Structural determinants of D-cycloserine efficacy at the NR1/NR2C NMDA receptors. J. Neurosci. 2010, 30, 2741–2754. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U. N-Methyl-D-aspartate (NMDA) receptor-based treatment approaches in schizophrenia: The first decade. Int. J. Neuropsychopharmacol. 2000, 3, 243–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, G.; Lane, H.-Y.; Yang, P.; Chong, M.-Y.; Lange, N. Glycine transporter I inhibitor, N-Methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 2004, 55, 452–456. [Google Scholar] [CrossRef]
- Lane, H.-Y.; Liu, Y.-C.; Huang, C.-L.; Chang, Y.-C.; Liau, C.-H.; Perng, C.-H.; Tsai, G.E. Sarcosine (N-Methylglycine) Treatment for Acute Schizophrenia: A Randomized, Double-Blind Study. Biol. Psychiatry 2008, 63, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Strzelecki, D.; Urban-Kowalczyk, M.; Wysokiński, A. Serum levels of interleukin 6 in schizophrenic patients during treatment augmentation with sarcosine (results of the PULSAR study). Hum. Psychopharmacol. Clin. Exp. 2018, 33, e2652. [Google Scholar] [CrossRef]
- Strzelecki, D.; Podgórski, M.; Kałużyńska, O.; Gawlik-Kotelnicka, O.; Stefańczyk, L.; Kotlicka-Antczak, M.; Gmitrowicz, A.; Grzelak, P. Supplementation of Antipsychotic Treatment with the Amino Acid Sarcosine Influences Proton Magnetic Resonance Spectroscopy Parameters in Left Frontal White Matter in Patients with Schizophrenia. Nutrients 2015, 7, 8767–8782. [Google Scholar] [CrossRef] [Green Version]
- Strzelecki, D.; Podgórski, M.; Kałużyńska, O.; Gawlik-Kotelnicka, O.; Stefańczyk, L.; Kotlicka-Antczak, M.; Gmitrowicz, A.; Grzelak, P. Supplementation of antipsychotic treatment with sarcosine—GlyT1 inhibitor—Causes changes of glutamatergic 1NMR spectroscopy parameters in the left hippocampus in patients with stable schizophrenia. Neurosci. Lett. 2015, 606, 7–12. [Google Scholar] [CrossRef]
- Umbricht, D.; Alberati, D.; Martin-Facklam, M.; Borroni, E.; Youssef, E.A.; Ostland, M.; Wallace, T.L.; Knoflach, F.; Dorflinger, E.; Wettstein, J.G.; et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: A randomized, double-blind, proof-of-concept study. JAMA Psychiatry 2014, 71, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugarski-Kirola, D.; Iwata, N.; Sameljak, S.; Reid, C.; Blaettler, T.; Millar, L.; Marques, T.R.; Garibaldi, G.; Kapur, S. Efficacy and safety of adjunctive bitopertin versus placebo in patients with suboptimally controlled symptoms of schizophrenia treated with antipsychotics: Results from three phase 3, randomised, double-blind, parallel-group, placebo-controlled, multicentre studies in the SearchLyte clinical trial programme. Lancet Psychiatry 2016, 3, 1115–1128. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Nolan, K.A.; Epstein, M.; Lehrfeld, N.; Shope, C.; Petkova, E.; Javitt, D.C. Neurophysiological Effects of Bitopertin in Schizophrenia. J. Clin. Psychopharmacol. 2017, 37, 447–451. [Google Scholar] [CrossRef]
- Fleischhacker, W.W.; Podhorna, J.; Gröschl, M.; Hake, S.; Zhao, Y.; Huang, S.; Keefe, R.S.E.; Desch, M.; Brenner, R.; Walling, D.P.; et al. Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: A double-blind, randomised, placebo-controlled phase 2 study. Lancet Psychiatry 2021, 8, 191–201. [Google Scholar] [CrossRef]
- Cain, C.K.; McCue, M.; Bello, I.; Creedon, T.; Tang, D.-I.; Laska, E.; Goff, D.C. D-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr. Res. 2014, 153, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, E.J.; Szilagyi, S.; Schwartz, M.P.; Bugarski-Kirola, D.; Kunzova, A.; Negi, S.; Stephanides, M.; Efferen, T.R.; Angrist, B.; Peselow, E.; et al. Effects of d-cycloserine on negative symptoms in schizophrenia. Schizophr. Res. 2004, 71, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Henderson, D.C.; Evins, A.; Amico, E. A placebo-controlled crossover trial of d-cycloserine added to clozapine in patients with schizophrenia. Biol. Psychiatry 1999, 45, 512–514. [Google Scholar] [CrossRef]
- Goff, D.C.; Tsai, G.; Levitt, J.; Amico, E.; Manoach, D.; Schoenfeld, D.A.; Hayden, D.L.; McCarley, R.; Coyle, J.T. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch. Gen. Psychiatry 1999, 56, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Goff, D.C.; Herz, L.; Posever, T.; Shih, V.; Tsai, G.; Henderson, D.C.; Freudenreich, O.; Evins, A.E.; Yovel, I.; Zhang, H.; et al. A six-month, placebo-controlled trial of d-cycloserine co-administered with conventional antipsychotics in schizophrenia patients. Psychopharmacologia 2004, 179, 144–150. [Google Scholar] [CrossRef]
- Goff, D.C.; Cather, C.; Gottlieb, J.D.; Evins, A.E.; Walsh, J.; Raeke, L.; Otto, M.W.; Schoenfeld, D.; Green, M.F. Once-weekly d-cycloserine effects on negative symptoms and cognition in schizophrenia: An exploratory study. Schizophr. Res. 2008, 106, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Heresco-Levy, U.; Ermilov, M.; Shimoni, J.; Shapira, B.; Silipo, G.; Javitt, D.C. Placebo-controlled trial of D-cycloserine added to conventional neuroleptics, olanzapine, or risperidone in schizophrenia. Am. J. Psychiatry 2002, 159, 480–482. [Google Scholar] [CrossRef]
- Rosse, R.B.; Fay-Mccarthy, M.; Kendrick, K.; Davis, E.R.; Deutsch, S.I. D-Cycloserine Adjuvant Therapy to Molindone in the Treatment of Schizophrenia. Clin. Neuropharmacol. 1996, 19, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, K.; Uezato, A.; Itasaka, M.; Atsuta, H.; Narushima, K.; Yamamoto, N.; Kurumaji, A.; Tomita, M.; Oshima, K.; Shoda, K.; et al. Association of schizophrenia onset age and white matter integrity with treatment effect of D-cycloserine: A randomized placebo-controlled double-blind crossover study. BMC Psychiatry 2017, 17, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafra, F.; Aragon, C.; Olivares, L.; Danbolt, N.; Gimenez, C.; Storm-Mathisen, J. Glycine transporters are differentially expressed among CNS cells. J. Neurosci. 1995, 15, 3952–3969. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Gonzalez-Gonzalez, I.M.; Gimenez, C.; Zafra, F. The scaffolding protein PSD-95 interacts with the glycine transporter GLYT1 and impairs its internalization. J. Neurochem. 2005, 95, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Betz, H.; Gomeza, J.; Armsen, W.; Scholze, P.; Eulenburg, V. Glycine transporters: Essential regulators of synaptic transmission. Biochem. Soc. Trans. 2006, 34 Pt 1, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Gorfinkel, Y.; Halman, S.; Lowe, J.A.; Periyalwar, P.; Schmidt, C.J.; Bergeron, R. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J. Physiol. 2004, 557, 489–500. [Google Scholar] [CrossRef]
- Vargas-Medrano, J.; Castrejon-Tellez, V.; Plenge, F.; Ramirez, I.; Miranda, M. PKCβ-dependent phosphorylation of the glycine transporter. Neurochem. Int. 2011, 59, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Gomeza, J.; Hülsmann, S.; Ohno, K.; Eulenburg, V.; Szöke, K.; Richter, D.; Betz, H. Inactivation of the Glycine Transporter 1 Gene Discloses Vital Role of Glial Glycine Uptake in Glycinergic Inhibition. Neuron 2003, 40, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Kurolap, A.; Hershkovitz, T.; Baris, H.N.; Adam, M.P.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. (Eds.) GLYT1 Encephalopathy, in GeneReviews(®®); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gabernet, L.; Pauly-Evers, M.; Schwerdel, C.; Lentz, M.; Bluethmann, H.; Vogt, K.; Alberati, D.; Mohler, H.; Boison, D. Enhancement of the NMDA receptor function by reduction of glycine transporter-1 expression. Neurosci. Lett. 2004, 373, 79–84. [Google Scholar] [CrossRef]
- Bergeron, R.; Meyer, T.M.; Coyle, J.T.; Greene, R.W. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc. Natl. Acad. Sci. USA 1998, 95, 15730–15734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberati, D.; Moreau, J.-L.; Lengyel, J.; Hauser, N.; Mory, R.; Borroni, E.; Pinard, E.; Knoflach, F.; Schlotterbeck, G.; Hainzl, D.; et al. Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology 2012, 62, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Kinney, G.G.; Sur, C.; Burno, M.; Mallorga, P.J.; Williams, J.B.; Figueroa, D.J.; Wittmann, M.; Lemaire, W.; Conn, P.J. The glycine transporter type 1 inhibitor N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine potentiates NMDA receptor-mediated responses in vivo and produces an antipsychotic profile in rodent behavior. J. Neurosci. 2003, 23, 7586–7591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depoortère, R.; Dargazanli, G.; Estenne-Bouhtou, G.; Coste, A.; Lanneau, C.; Desvignes, C.; Poncelet, M.; Héaulme, M.; Santucci, V.; Decobert, M.; et al. Neurochemical, Electrophysiological and Pharmacological Profiles of the Selective Inhibitor of the Glycine Transporter-1 SSR504734, a Potential New Type of Antipsychotic. Neuropsychopharmacology 2005, 30, 1963–1985. [Google Scholar] [CrossRef]
- Fone, K.C.; Watson, D.J.; Billiras, R.I.; Sicard, D.I.; Dekeyne, A.; Rivet, J.-M.; Gobert, A.; Millan, M.J. Comparative Pro-cognitive and Neurochemical Profiles of Glycine Modulatory Site Agonists and Glycine Reuptake Inhibitors in the Rat: Potential Relevance to Cognitive Dysfunction and Its Management. Mol. Neurobiol. 2020, 57, 2144–2166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Hyrc, K.; Thio, L.L. The glycine transport inhibitor sarcosine is an NMDA receptor co-agonist that differs from glycine. J. Physiol. 2009, 587, 3207–3220. [Google Scholar] [CrossRef]
- Zhang, H.X.; Lyons-Warren, A.; Thio, L.L. The glycine transport inhibitor sarcosine is an inhibitory glycine receptor agonist. Neuropharmacology 2009, 57, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Kopec, K.; Flood, D.G.; Gasior, M.; McKenna, B.A.W.; Zuvich, E.; Schreiber, J.; Salvino, J.M.; Durkin, J.T.; Ator, M.A.; Marino, M.J. Glycine transporter (GlyT1) inhibitors with reduced residence time increase prepulse inhibition without inducing hyperlocomotion in DBA/2 mice. Biochem. Pharmacol. 2010, 80, 1407–1417. [Google Scholar] [CrossRef]
- Harvey, P.D.; Bowie, C.R.; McDonald, S.; Podhorna, J. Evaluation of the Efficacy of BI 425809 Pharmacotherapy in Patients with Schizophrenia Receiving Computerized Cognitive Training: Methodology for a Double-blind, Randomized, Parallel-group Trial. Clin. Drug Investig. 2020, 40, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Desch, M.; Wunderlich, G.; Goettel, M.; Goetz, S.; Liesenfeld, K.-H.; Chan, T.S.; Rosenbrock, H.; Sennewald, R.; Link, J.; Keller, S.; et al. Effects of Cytochrome P450 3A4 Induction and Inhibition on the Pharmacokinetics of BI 425809, a Novel Glycine Transporter 1 Inhibitor. Eur. J. Drug Metab. Pharmacokinet. 2021, 47, 91–103. [Google Scholar] [CrossRef]
- Wolinsky, E. Statement of the Tuberculosis Committee of the Infectious Diseases Society of America. Clin. Infect. Dis. 1993, 16, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Smits, J.A.; Rosenfield, D.; Otto, M.W.; Marques, L.; Davis, M.L.; Meuret, A.E.; Simon, N.M.; Pollack, M.H.; Hofmann, S.G. D-cycloserine enhancement of exposure therapy for social anxiety disorder depends on the success of exposure sessions. J. Psychiatr. Res. 2013, 47, 1455–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, L.J.; Waters, A.M.; Tiralongo, E.; Mathieu, S.; McKenzie, M.; Garbharran, V.; Ware, R.S.; Zimmer-Gembeck, M.J.; McConnell, H.; Lavell, C.; et al. Efficacy of D-cycloserine augmented brief intensive cognitive-behavioural therapy for paediatric obsessive-compulsive disorder: A randomised clinical trial. Depress. Anxiety 2022. [Google Scholar] [CrossRef] [PubMed]
- Kvale, G.; Hansen, B.; Hagen, K.; Abramowitz, J.S.; Børtveit, T.; Craske, M.G.; Franklin, M.E.; Haseth, S.; Himle, J.A.; Hystad, S.; et al. Effect of D-Cycloserine on the Effect of Concentrated Exposure and Response Prevention in Difficult-to-Treat Obsessive-Compulsive Disorder: A Randomized Clinical Trial. JAMA Netw. Open. 2020, 3, e2013249. [Google Scholar] [CrossRef] [PubMed]
- Olden, M.; Wyka, K.; Cukor, J.; Peskin, M.; Altemus, M.; Lee, F.S.; Finkelstein-Fox, L.; Rabinowitz, T.; Difede, J. Pilot Study of a Telehealth-Delivered Medication-Augmented Exposure Therapy Protocol for PTSD. J. Nerv. Ment. Dis. 2017, 205, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.; Selby, B.; Ismail, Z.; McGirr, A. D-cycloserine normalizes long-term motor plasticity after transcranial magnetic intermittent theta-burst stimulation in major depressive disorder. Clin. Neurophysiol. 2021, 132, 1770–1776. [Google Scholar] [CrossRef]
- Rothbaum, B.O.; Price, M.; Jovanovic, T.; Norrholm, S.D.; Gerardi, M.; Dunlop, B.; Davis, M.; Bradley, B.; Duncan, E.J.; Rizzo, A.; et al. A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am. J. Psychiatry 2014, 171, 640–648. [Google Scholar] [CrossRef]
- Mataix-Cols, D.; Fernández de la Cruz, L.; Monzani, B.; Rosenfield, D.; Andersson, E.; Pérez-Vigil, A.; Frumento, P.; de Kleine, R.A.; Difede, J.; Dunlop, B.W.; et al. D-Cycloserine Augmentation of Exposure-Based Cognitive Behavior Therapy for Anxiety, Obsessive-Compulsive, and Posttraumatic Stress Disorders: A Systematic Review and Meta-analysis of Individual Participant Data. JAMA Psychiatry 2017, 74, 501–510. [Google Scholar] [CrossRef]
- Polese, D.; De Serpis, A.A.; Ambesi-Impiombato, A.; Muscettola, G.; De Bartolomeis, A. Homer 1a Gene Expression Modulation by Antipsychotic Drugs Involvement of the Glutamate Metabotropic System and Effects of D-Cycloserine. Neuropsychopharmacology 2002, 27, 906–913. [Google Scholar] [CrossRef]
- Van Berckel, B.N.; Evenblij, C.N.; van Loon, B.J.; Maas, M.F.; van der Geld, M.A.; Wynne, H.J.; van Ree, J.M.; Kahn, R.S. D-cycloserine increases positive symptoms in chronic schizophrenic patients when administered in addition to antipsychotics: A double-blind, parallel, placebo-controlled study. Neuropsychopharmacology 1999, 21, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Kuppili, P.P.; Menon, V.; Sathyanarayanan, G.; Sarkar, S.; Andrade, C. Efficacy of adjunctive d-Cycloserine for the treatment of schizophrenia: A systematic review and meta-analysis of randomized controlled trials. J. Neural Transm. 2021, 128, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Hitachi, K.; Okamoto, H.; Saitoh, M.; Odagiri, M.; Ohfusa, R.; Shimada, T.; Taguchi, A.; Taniguchi, A.; Tsuchida, K.; et al. Development of Myostatin Inhibitory d-Peptides to Enhance the Potency, Increasing Skeletal Muscle Mass in Mice. ACS Med. Chem. Lett. 2022, 13, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Aillaud, I.; Kaniyappan, S.; Chandupatla, R.R.; Ramirez, L.M.; Alkhashrom, S.; Eichler, J.; Horn, A.H.C.; Zweckstetter, M.; Mandelkow, E.; Sticht, H.; et al. A novel D-amino acid peptide with therapeutic potential (ISAD1) inhibits aggregation of neurotoxic disease-relevant mutant Tau and prevents Tau toxicity in vitro. Alzheimer’s Res. Ther. 2022, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.M.; Mayr, L.M.; Minor, D.L.; Milhollen, M.A.; Burgess, M.W.; Kim, P.S. Identification of d-Peptide Ligands Through Mirror-Image Phage Display. Science 1996, 271, 1854–1857. [Google Scholar] [CrossRef] [Green Version]
- Malhis, M.; Kaniyappan, S.; Aillaud, I.; Chandupatla, R.R.; Ramirez, L.M.; Zweckstetter, M.; Horn, A.H.C.; Mandelkow, E.; Sticht, H.; Funke, S.A. Potent Tau Aggregation Inhibitor D-Peptides Selected against Tau-Repeat 2 Using Mirror Image Phage Display. ChemBioChem 2021, 22, 3049–3059. [Google Scholar] [CrossRef]
- Sadowski, M.; Pankiewicz, J.; Scholtzova, H.; Ripellino, J.A.; Li, Y.; Schmidt, S.D.; Mathews, P.M.; Fryer, J.D.; Holtzman, D.M.; Sigurdsson, E.M.; et al. A Synthetic Peptide Blocking the Apolipoprotein E/β-Amyloid Binding Mitigates β-Amyloid Toxicity and Fibril Formation in Vitro and Reduces β-Amyloid Plaques in Transgenic Mice. Am. J. Pathol. 2004, 165, 937–948. [Google Scholar] [CrossRef]
- Chalifour, R.J.; McLaughlin, R.W.; Lavoie, L.; Morissette, C.; Tremblay, N.; Boulé, M.; Sarazin, P.; Stéa, D.; Lacombe, D.; Tremblay, P.; et al. Stereoselective Interactions of Peptide Inhibitors with the β-Amyloid Peptide. J. Biol. Chem. 2003, 278, 34874–34881. [Google Scholar] [CrossRef] [Green Version]
- Funke, S.A.; Van Groen, T.; Kadish, I.; Bartnik, D.; Nagel-Steger, L.; Brener, O.; Sehl, T.; Batra-Safferling, R.; Moriscot, C.; Schoehn, G.; et al. Oral Treatment with the d-Enantiomeric Peptide D3 Improves the Pathology and Behavior of Alzheimer’s Disease Transgenic Mice. ACS Chem. Neurosci. 2010, 1, 639–648. [Google Scholar] [CrossRef]
- Engel, H.; Guischard, F.; Krause, F.; Nandy, J.; Kaas, P.; Höfflin, N.; Köhn, M.; Kilb, N.; Voigt, K.; Wolf, S.; et al. finDr: A web server for in silico D-peptide ligand identification. Synth. Syst. Biotechnol. 2021, 6, 402–413. [Google Scholar] [CrossRef]
- Valiente, P.A.; Wen, H.; Nim, S.; Lee, J.; Kim, H.J.; Kim, J.; Perez-Riba, A.; Paudel, Y.P.; Hwang, I.; Kim, K.-D.; et al. Computational Design of Potent D-Peptide Inhibitors of SARS-CoV. J. Med. Chem. 2021, 64, 14955–14967. [Google Scholar] [CrossRef]
- He, W.; Zhang, Z.; Yang, W.; Zheng, X.; You, W.; Yao, Y.; Yan, J.; Liu, W. Turing milk into pro-apoptotic oral nanotherapeutic: De novo bionic chiral-peptide supramolecule for cancer targeted and immunological therapy. Theranostics 2022, 12, 2322–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Tan, Y.; Hu, R.; Niu, M.M. An NRP1/MDM2-Targeted D-Peptide Supramolecular Nanomedicine for High-Efficacy and Low-Toxic Liver Cancer Therapy. Adv. Healthc. Mater. 2021, 10, e2002197. [Google Scholar] [CrossRef]
- Porcelli, S.; Van Der Wee, N.; van der Werff, S.; Aghajani, M.; Glennon, J.C.; van Heukelum, S.; Mogavero, F.; Lobo, A.; Olivera, F.J.; Lobo, E.; et al. Social brain, social dysfunction and social withdrawal. Neurosci. Biobehav. Rev. 2018, 97, 10–33. [Google Scholar] [CrossRef] [PubMed]
- Azmanova, M.; Pitto-Barry, A.; Barry, N.P.E. Schizophrenia: Synthetic strategies and recent advances in drug design. MedChemComm 2018, 9, 759–782. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Conn, P.J. Metabotropic glutamate receptors as emerging targets for the treatment of schizophrenia. Mol. Pharmacol. 2022, 101, 275–285. [Google Scholar] [CrossRef]
- Jankowska, A.; Satała, G.; Partyka, A.; Wesołowska, A.; Bojarski, A.J.; Pawłowski, M.; Chłoń-Rzepa, G. Discovery and Development of Non-Dopaminergic Agents for the Treatment of Schizophrenia: Overview of the Preclinical and Early Clinical Studies. Curr. Med. Chem. 2019, 26, 4885–4913. [Google Scholar] [CrossRef]
- Gouvêa-Junqueira, D.; Falvella, A.C.B.; Antunes, A.; Seabra, G.; Brandão-Teles, C.; Martins-De-Souza, D.; Crunfli, F. Novel Treatment Strategies Targeting Myelin and Oligodendrocyte Dysfunction in Schizophrenia. Front. Psychiatry 2020, 11, 379. [Google Scholar] [CrossRef]
- Hashimoto, K. Recent Advances in the Early Intervention in Schizophrenia: Future Direction from Preclinical Findings. Curr. Psychiatry Rep. 2019, 21, 75. [Google Scholar] [CrossRef]
- Sonnenschein, S.F.; Grace, A.A. Emerging therapeutic targets for schizophrenia: A framework for novel treatment strategies for psychosis. Expert Opin. Ther. Targets 2020, 25, 15–26. [Google Scholar] [CrossRef]
- Roychaudhuri, R.; Snyder, S.H. Mammalian D-cysteine: A novel regulator of neural progenitor cell proliferation: Endogenous D-cysteine, the stereoisomer with rapid spontaneous in vitro racemization rate, has major neural roles. Bioessays 2022, 44, e2200002. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, K.; Fu, Z.; Yu, D.; Huang, H.; Zang, X.; Mo, X. Brain Development and Akt Signaling: The Crossroads of Signaling Pathway and Neurodevelopmental Diseases. J. Mol. Neurosci. 2016, 61, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-C.; Gao, W.-J. GSK-3β activity and hyperdopamine-dependent behaviors. Neurosci. Biobehav. Rev. 2011, 35, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunii, Y.; Matsumoto, J.; Izumi, R.; Nagaoka, A.; Hino, M.; Shishido, R.; Sainouchi, M.; Akatsu, H.; Hashizume, Y.; Kakita, A.; et al. Evidence for Altered Phosphoinositide Signaling-Associated Molecules in the Postmortem Prefrontal Cortex of Patients with Schizophrenia. Int. J. Mol. Sci. 2021, 22, 8280. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.-Z.; Chang, C.-Y.; Huang, T.-R.; Studer, V.; Wang, T.-W.; Lai, W.-S. Lithium for schizophrenia: Supporting evidence from a 12-year, nationwide health insurance database and from Akt1-deficient mouse and cellular models. Sci. Rep. 2020, 10, 647. [Google Scholar] [CrossRef] [Green Version]
- Moretti, P.N.; Ota, V.K.; Gouvea, E.S.; Pedrini, M.; Santoro, M.L.; Talarico, F.; Spindola, L.M.; Carvalho, C.M.; Noto, C.; Xavier, G.; et al. Accessing Gene Expression in Treatment-Resistant Schizophrenia. Mol. Neurobiol. 2018, 55, 7000–7008. [Google Scholar] [CrossRef]
- Rampino, A.; Marakhovskaia, A.; Soares-Silva, T.; Torretta, S.; Veneziani, F.; Beaulieu, J.M. Antipsychotic Drug Responsiveness and Dopamine Receptor Signaling; Old Players and New Prospects. Front. Psychiatry 2019, 9, 702. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Banaclocha, M. N-acetyl-cysteine in Schizophrenia: Potential Role on the Sensitive Cysteine Proteome. Curr. Med. Chem. 2020, 27, 6424–6439. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, Y.; Chen, Z.; Kumata, K.; Van, R.; Rong, J.; Shao, T.; Hatori, A.; Mori, W.; Yu, Q.; et al. Synthesis and preliminary evaluation of 4-hydroxy-6-(3-[11C]methoxyphenethyl)pyridazin-3(2H)-one, a 11C-labeled-amino acid oxidase (DAAO) inhibitor for PET imaging. Bioorganic Med. Chem. Lett. 2020, 30, 127326. [Google Scholar] [CrossRef]





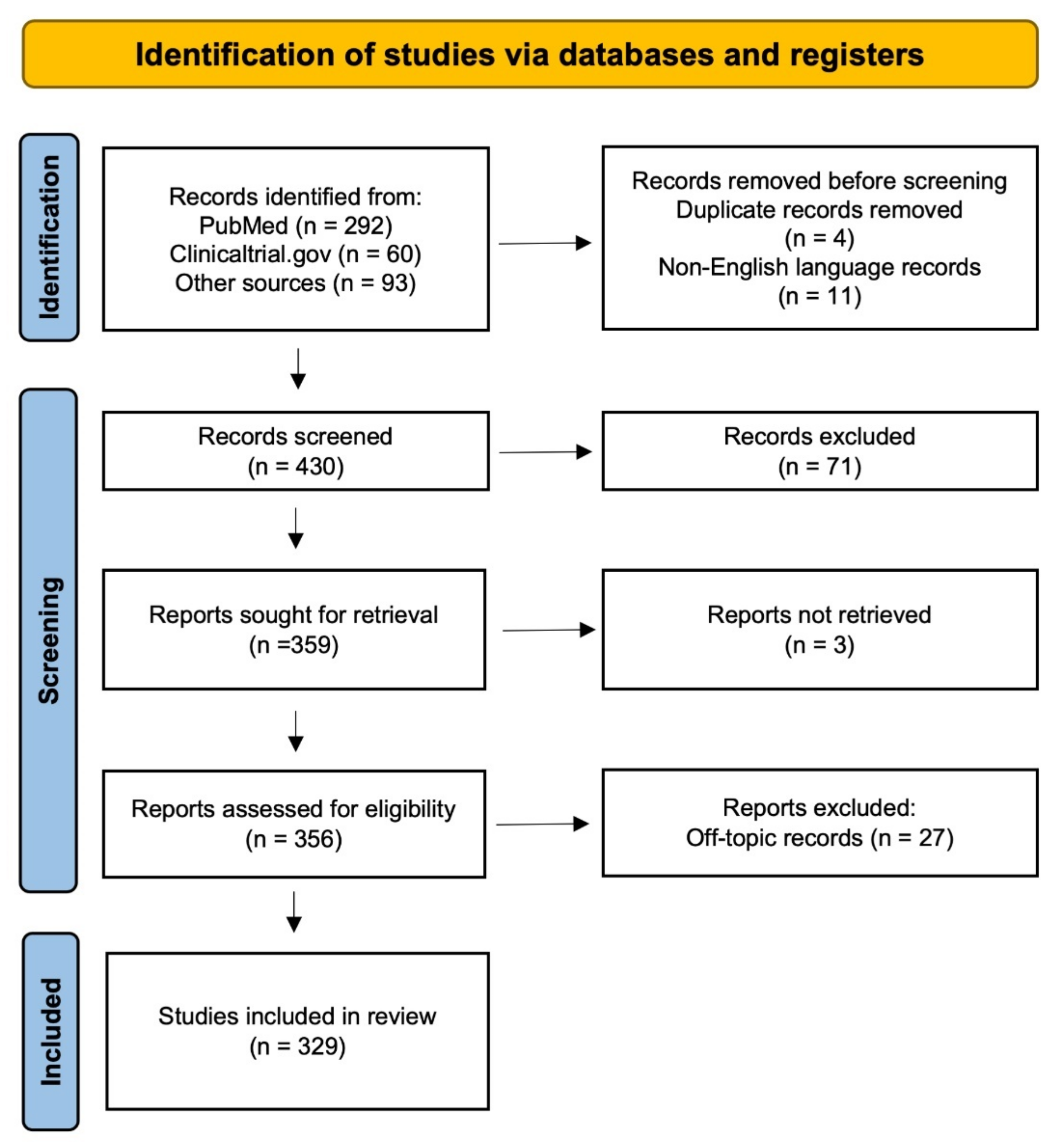{kind=link}
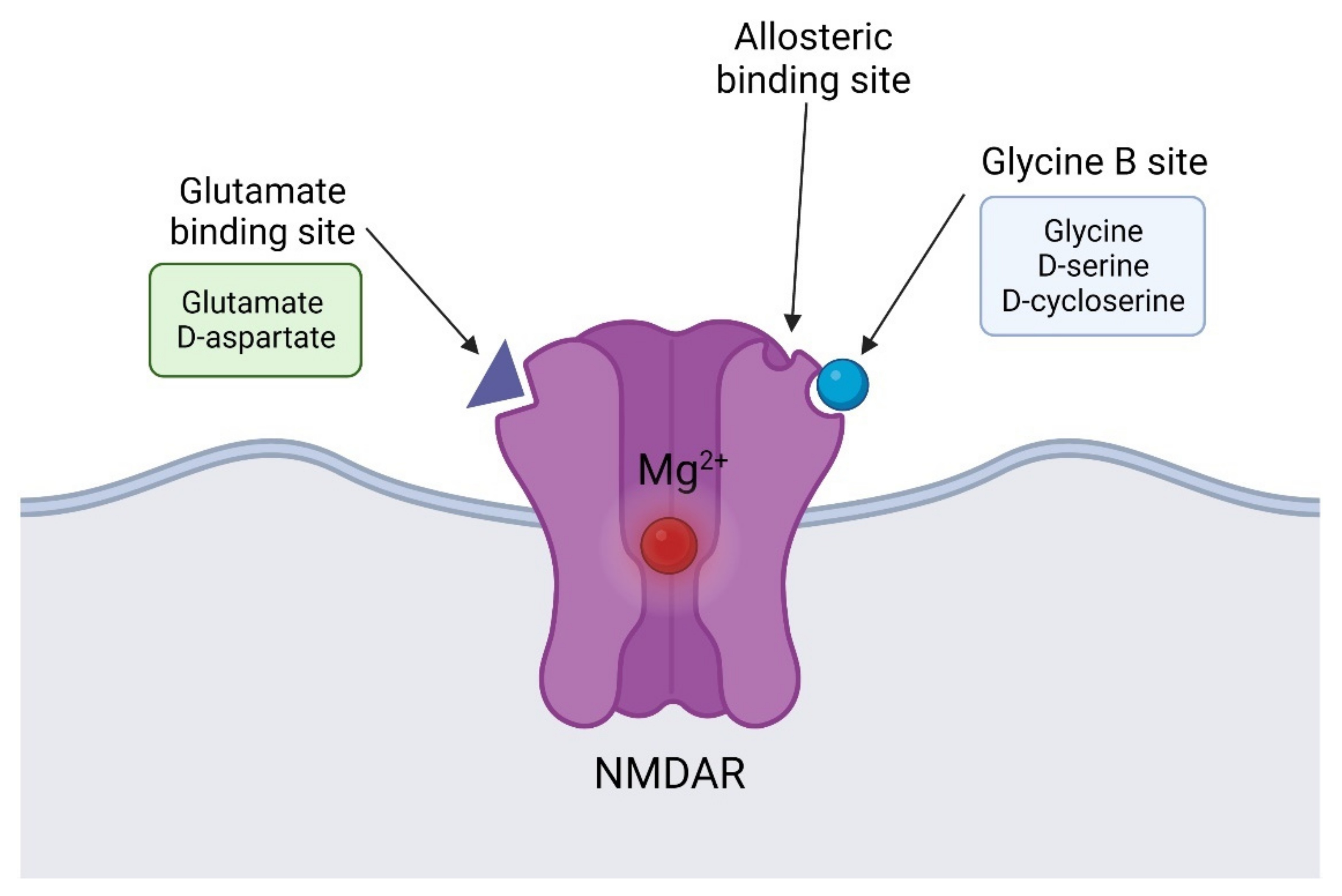{kind=link}
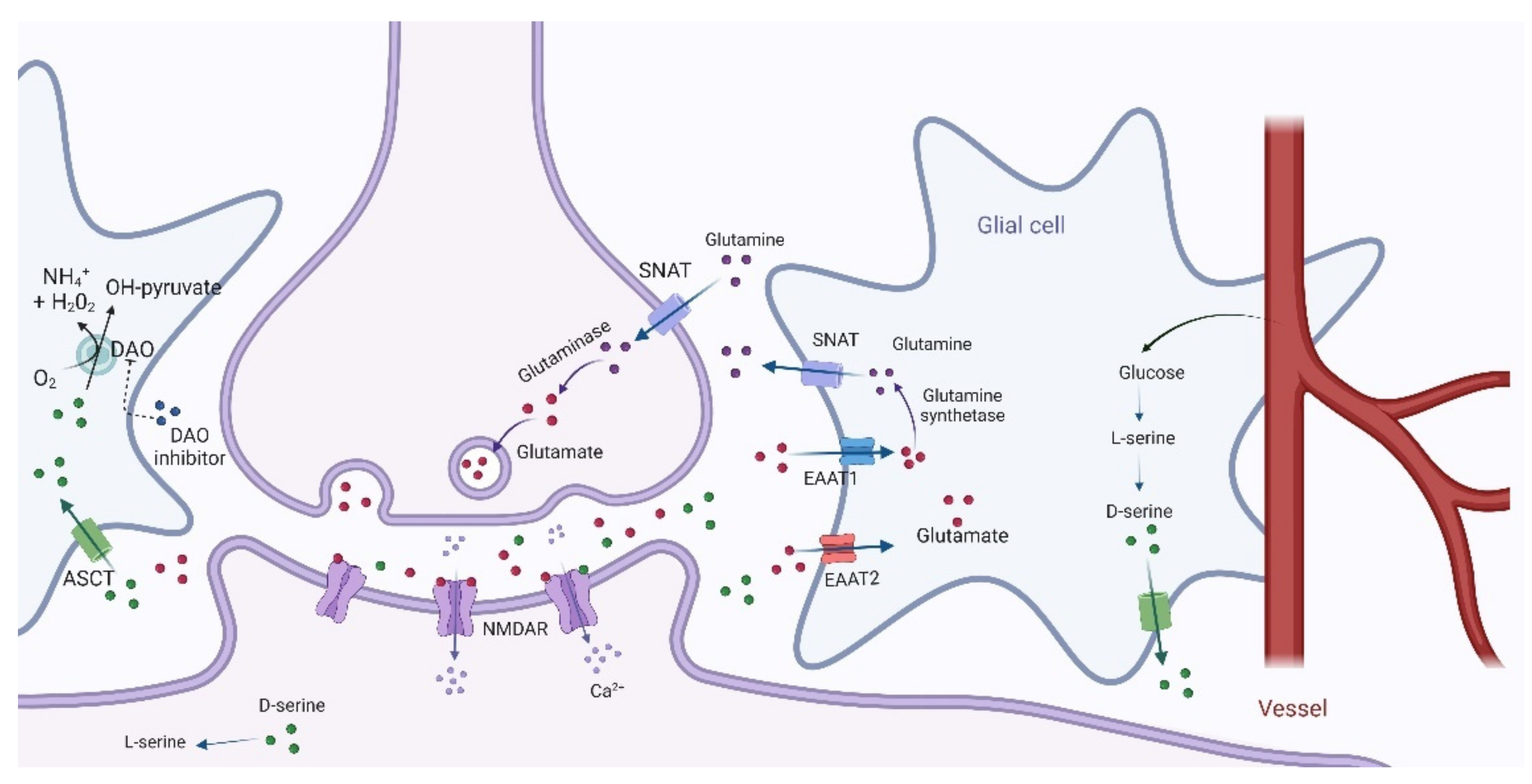{kind=link}
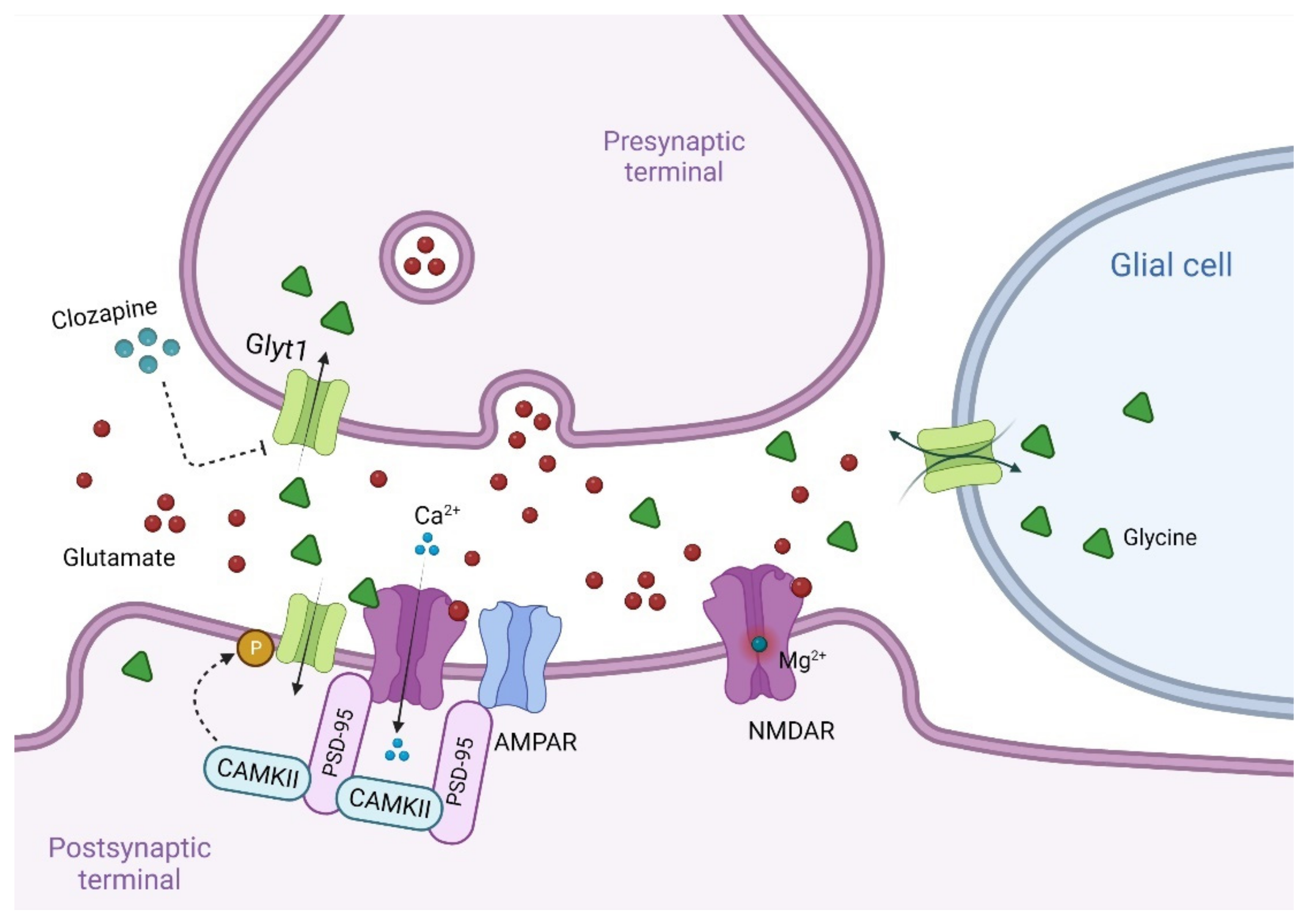{kind=link}
| Agent Added | Author | Study Design | Groups of Treatment | Mean Age | n | Dose | Duration | Stable Antipsychotic Regimens | Outcome | Side Effects | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D-serine | Kantrowitz et al., 2018 [223] | Double-blind crossover trial | D-serine | 40 ± 11 | 14 | 60 mg/kg/day | 6 weeks | CPZ equivalents (mg): 965 ± 760 | Improvement in total PANSS symptoms Reduction in evoked power for the α band | - | |
| Placebo | - | ||||||||||
| Weiser et al., 2012 [228] | RCT | D-serine | Not retrieved | 195 | 2 g/day | - | Not retrieved | No significant differences in SANS and MATRICS battery score | - | ||
| Placebo | |||||||||||
| Tsai et al., 1998 [226] | Double-blind, placebo-controlled trial | D-serine | 31.7 ± 7.5 | 15 | 30 mg/kg/day | 6 weeks | Sulpiride (n = 6), Haloperidol (n = 5); Risperidone (n = 4); Pipotiazine (n = 2) Fluphenthixol (n = 2) Thiothixene (n = 1) Pipotiazine (n = 2) Haloperidol and pipotiazine (n = 1) Pipotiazine and chlorpromazine (n = 1) Fluphenazine and chlorpromazine (n = 1) Risperidone and chlorpromazine (n = 1) Trifluoperazine and chlorpromazine (n = 1) Trifluoperazine and haloperidol (n = 1) One patient was antipsychotic free. | Improvement in PANSS positive SANS PANSS-cognitive WCST CGI | No significant side effects | ||
| Placebo | 33.9 ± 6.6 | 14 | - | ||||||||
| Heresco-Levy et al., 2005 [222] | double-blind, placebo-controlled trial | Risperidone + D-serine Olanzapine + D-serine | 42.7 ± 13.1 | 21 | 30 mg/kg/day | 6 weeks | - | Improvement in BPRS SANS PANSS (negative, positive, cognitive) | No significant clinical or laboratory side effects | ||
| Risperidone + Placebo Olanzapine + Placebo | 47.4 ± 14.2 | 18 | |||||||||
| Lane et al., 2005 [224] | RCT | D-serine | 31.8 ± 10.2 | 21 | 2 g/day | 6 weeks | Risperidone | No significant differences | Weight gain (67%) Palpitation (42%) | ||
| Placebo | 34.1 ± 8.7 | 23 | |||||||||
| Lane et al., 2010 [225] | RCT | D-serine | 30.7 ± 9.6 | 20 | 2 g/day | 6 weeks | Risperidone Olanzapine Quetiapine | No significant differences | No significant side effects | ||
| Placebo | 31.5 ± 7.9 | 20 | |||||||||
| Tsai et al., 2010 [227] | RCT | D-serine | 42.6 ± 3.6 | 10 | 30 mg/kg/day | 6 weeks | Clozapine mean dose: 363 ± 128 | No significant differences | No differences in side effects | ||
| Placebo | 39.5 ± 5.5 | 10 | Clozapine mean dose: 315 ± 146 | ||||||||
| Kantrowitz et al., 2010 [230] | Open label trial | D-serine low dose | 41.7 ± 11.4 | 12 | 30 mg/kg | CPZ Equivalents/day | 468.8 ± 252 | Improvement in PANSS and CGI | - | ||
| D-serine mild dose | 43.5 ± 9.4 | 19 | 60 mg/kg | 602.6 ± 295 | - | ||||||
| D-serine high dose | 43.2 ± 9.6 | 16 | 120 mg/kg | 493.7 ± 319 | Nephrotoxic-like pattern at 120 mg/kg which resolved upon D-serine discontinuation (6.25%) Asymptomatic transaminitis (12.5%) Insomnia after one dose and GI distress (12.5%) | ||||||
| Kantrowitz et al., 2015 [26] | Double-blind, placebo-controlled trial | D-serine | 13–35 | 15 | 60 mg/kg/day | 16 weeks | - | Scale of Prodromal Symptoms negative score | - | ||
| Placebo | 20 | - | |||||||||
| D-alanine | Tsai et al., 2006 [27] | RCT | D-alanine | 30.9 ± 6.5 | 14 | 100 mg/kg daily | 6 weeks | CPZ equivalents (mg): 468 ± 478 | Sulpiride; Haloperidol; Risperidone; Pipotiazine; Pipotiazine and chlorpromazine; Pipotiazine and sulpiride; Thioridazine; Trifluoperazine and chlorpromazine; Fluphenazine decanoate and chlorpromazine | Improvement in PANSS-positive PANSS-cognitive and SANS score | No significant differences |
| Placebo | 31.8 ± 7.4 | 18 | CPZ equivalents (mg): 364 ± 220 | ||||||||
| Authors | Groups of Treatment | Mean Age | n | Dose | Duration | Stable Antipsychotic Regimens | Outcome | Side Effects |
|---|---|---|---|---|---|---|---|---|
| Lane et al., 2013 [317] | Sodium benzoate | 38.4 ± 9.7 | 25 | 1 g/day | 6 weeks | Amisulpride Chlorpromazine Flupenthixol Haloperidol Quetiapine fumarate Risperidone Sulpiride Ziprasidone Zotepine | The sodium benzoate group exhibited better performances in SANS, GAF, QOLS, CGI, HDRS, speed of processing, visual learning and memory | No significant differences were found between the two groups in Simpson-Angus Rating Scale, AIMS, and Barnes Akathisia Scale scores |
| Placebo | 36.3 ± 7.9 | 27 | - | |||||
| Scott et al., 2020 [318] | Sodium benzoate | 21.7 ± 4.7 | 49 | 1 g/day | 12 weeks | Aripiprazole Amisulpride Brexpiprazole Clozapine Haloperidol Olanzapine Lurasidone Paliperidone Quetiapine Risperidone | No improvements were found in the sodium benzoate group | No significant differences were found between groups |
| Placebo | 21.2 ± 3.4 | 50 | - | |||||
| Lin et al., 2018 [311] | Sodium benzoate | 44.3 ± 7.2 | 20 | 1 g/day | 6 weeks | Clozapine | Improvements in negative and overall symptomatology and quality of life were detected in sodium benzoate groups compared to placebo | No significant differences were found between the two groups in Simpson-Angus Rating Scale, AIMS, and Barnes Akathisia Scale scores |
| Sodium benzoate | 44.8 ± 8.1 | 20 | 2 g/day | |||||
| Placebo | 47 ± 11.9 | 20 | - | |||||
| Lin et al., 2017 [312] | Sarcosine + sodium benzoate | 37.8 ± 9.6 | 21 | 2 g/day + 1 g/day | 12 weeks | Amisulpride Aripiprazole Olanzapine Paliperidone Quetiapine Risperidone Zotepine | Improvement in GAF and cognitive battery compared to sarcosine group | - |
| Sarcosine | 38.2 ± 9.3 | 21 | 2 g/day | Improvement in reasoning and problem solving compared to placebo | ||||
| Placebo | 39.1 ± 9.5 | 21 | - | - |
| Agent Added | Author | Study Design | Groups of Treatment | Mean Age | n | Doses | Duration | Stable Antipsychotic Regimens | Outcome | Side Effects |
|---|---|---|---|---|---|---|---|---|---|---|
| Glycine | Serrita et al., 2019 [334] | RCT | Glycine | 48.60 ± 5.01 | 10 | 0.8 g/kg | 12 weeks | Antipsychotics | No significant differences in PANSS score | No significant differences |
| Placebo | 49.20 ± 4.84 | 10 | ||||||||
| Rosse et al., 1989 [345] | Open-label Pilot study | Glycine | 38 ± 15 | 6 | 18.8 g/day | 8 weeks | Haloperidol; Benztropine; Thiothixene; Vitamin E; Loxapine | Beneficial effects in two patients at SANS scale. Two others are worsened | Reduction in neuroleptic-induced muscle stiffness and extrapyramidal dysfunction in three patients. | |
| Potkin et al., 1999 [335] | RCT | Glycine | 12.4 ± 7.2 | 9 | 30 mg/day | 12 weeks | Clozapine (400–1200 mg/day) | No significant treatment effects | No significant differences | |
| Placebo | 10 | Improvement in BPRS positive symptoms | ||||||||
| Javitt et al., 1994 [336] | RCT | Glycine | - | 14 | 30 mg/day | 8 weeks | Unknown | Improvement in PANSS negative symptoms domain | - | |
| Placebo | ||||||||||
| Heresco-Levy et al., 1999 [339] | RCT | Glycine | 38.8 ± 11.0 | 9 | 61.2 ± 13.4 | 6 weeks | Clozapine mean dose: 471.0 ± 207.8 mg Chlorpromazine equivalents: 240.0 ± 146.3 | Significant increase in glycine and serine levels Significant improvement in PANSS—negative symptoms and BPRS total score | No significant differences | |
| Placebo | 10 | |||||||||
| Buchanan et al., 2007 [343] | RCT | Glycine | 42.6 ± 10.8 | 42 | 60 g/day | 16 weeks | Unknown | non-significant differences in SANS score Significant improvement in CGI score in D-cycloserine group compared to placebo | Worsened nausea and dry mouth in glycine group compared to placebo | |
| D-cycloserine | 44.4 ± 10.4 | 46 | 50 g/day | |||||||
| Placebo | 43.4 ± 11.4 | 45 | - | |||||||
| Costa et al., 1990 [344] | Open-label Pilot study | Glycine | 34.8 | 11 | 15 g/day | 5 weeks | Increasing amount in second generation antipsychotics | Decrease in BPRS score in 33% of patients | Upper gastrointestinal discomfort in 17% of patients | |
| Liederman et al., 1996 [346] | Open-label Pilot study | Glycine | 45.0 ± 7.6 | 5 | 60.0 g/day | 8 weeks | Clozapine (2) Risperidone (2) Haloperidol (1) | Significant improvement in negative symptoms was found using the SANS | No adverse effects; Reduction in EPS | |
| Heresco-Levy et al., 1996 [338] | RCT | Glycine | 41.36 ± 12.93 | 11 | 60.63 ± 12.98 | 6 weeks | Clozapine (4) Thioridazine (3) Haloperidol (2) Chlorpromazine (1) Perphenazine (1) | Improvement in PANSS-negative symptoms PANSS-general psychopathology and PANSS total score | No adverse effects | |
| Placebo | ||||||||||
| Javitt et al., 2001 [337] | RCT | Glycine | 39.6 ± 5.5 | 12 | 0.8 g/kg/day | 6 weeks | Olanzapine (6) Clozapine (4) Mesoridiazine (1) Haloperidol (1) | Improvement in PANSS- negative symptoms | No significant adverse effects | |
| Unknown | ||||||||||
| Placebo | ||||||||||
| Evins et al., 2000 [341] | RCT | Glycine | 39.0 ± 7.0 | 27 | 60 g/day | 8 weeks | Clozapine | No significant treatment effects | No significant adverse effects | |
| Placebo | ||||||||||
| Diaz et al., 2005 [342] | RCT | Glycine | 39.5 ± 12.44 | 6 | 60 g/day | 8 weeks | Clozapine mean dose: 575 mg/day | No significant treatment effects | Nausea and vomiting (16%) | |
| Placebo | 6 | - | ||||||||
| Greenwood et al., 2018 [340] | RCT | Glycine | 36.0 ± 7.8 | 12 | 24.8 g/day | 6 weeks | Unspecified antipsychotics medication | Improvement in PANSS-Total, PANSS-Negative and PANSS-General symptoms Improvement in MMN | - | |
| Placebo | 40.2 ± 8.9 | 10 | ||||||||
| Heresco-Levy et al., 2004 [326] | RCT | Glycine | 44.7 ± 10.8 | 7 | 55.2 ± 10.1 g/day | 6 weeks | Olanzapine (10) mean dose: 14.3 ± 6.2 mg/day Risperidone mean dose (4): 6.20 ± 3.08 mg/day | Improvement in five-factor PANSS | Decrease in EPS, upper gastrointestinal tract discomfort with nausea (12%) | |
| Placebo | 7 | |||||||||
| Sarcosine | Lane et al., 2005b [224] | RCT | Sarcosine | 36.1 ± 10.2 | 21 | 2 g/day | 6 weeks | Risperidone | Improvement in PANSS total, positive, and negative score; SANS-20 and SANS-17 | Treatment-emergent adverse events other than extrapyramidal symptoms are similar in the three groups |
| D-serine | 31.8 ± 10.4 | 21 | 2 g/day | - | ||||||
| Placebo | 34.1 ± 8.7 | 23 | - | - | ||||||
| Lane et al., 2006 [347] | RCT | Sarcosine | 36.7 ± 10.1 | 10 | 2 g/day | 6 weeks | Clozapine mean dose: 306 ± 159 mg | Non-significant differences | Non-significant differences | |
| Placebo | 35.5 ± 6.6 | 10 | Clozapine mean dose: 305 ± 55 mg | |||||||
| Lane et al., 2010 [225] | RCT | Sarcosine | 30.4 ± 10.6 | 20 | Not retrieved | 6 weeks | Risperidone Olanzapine Quetiapine | Not retrieved | - | |
| Placebo | 31.5 ± 7.9 | 20 | ||||||||
| D-serine | 30.7 ± 9.6 | 20 | ||||||||
| Lin et al., 2017 [312] | RCT | Sarcosine + sodium benzoate | 37.8 ± 9.6 | 21 | 2 g/day + 1 g/day | 12 weeks | Amisulpride Aripiprazole Olanzapine Paliperidone Quetiapine Risperidone Zotepine | Improvement in GAF and cognitive battery compared to sarcosine group | - | |
| Sarcosine | 38.2 ± 9.3 | 21 | 2 g/day | Improvement in reasoning and problem solving compared to placebo | ||||||
| Placebo | 39.1 ± 9.5 | 21 | - | - | ||||||
| Tsai et al., 2004 [350] | RCT | Sarcosine | 29.8 ± 7.2 | 17 | 2 g/day | 6 weeks | Chlorpromazine equivalence: 409 ± 320 | Improvement in PANSS score | No significant side effects | |
| Placebo | 33.4 ± 8.3 | 21 | Chlorpromazine equivalence: 433 ± 243 | |||||||
| Lane et al., 2008 [351] | RCT | Sarcosine | 31.3 ± 10.4 | 9 | 1 g/day | 6 weeks | Drug-free | No significant differences between groups | Insomnia, weight gain (1-2 kg), sedation, constipation, and fatigability. These effects were all mild and brief. | |
| Sarcosine | 34.3 ± 11.2 | 11 | 2 g/day | |||||||
| Strzelecki et al., 2018 [352] | RCT | Sarcosine | 37.3 ± 11.3 | 29 | 2 g/day | 6 months | First- and second-generation antipsychotics | Improvement in total, general, and negative PANSS scores | No significant side effects | |
| Placebo | 40.2 ± 10.1 | 30 | ||||||||
| Strzelecki et al., 2015 [353,354] | RCT | Sarcosine | 36.5 | 25 | 2 g/day | 6 months | Antipsychotics except clozapine | Improvement in total PANSS scores | No significant side effects | |
| Placebo | 40 | 25 | ||||||||
| Bitopertin | Umbricht et al., 2014 [355] | RCT | Bitopertin high dose | 38.9 ± 9.5 | 79 | 60 mg/day | 8 weeks | Aripiprazole Olanzapine Quetiapine Paliperidone Risperidone Risperidone long-acting injection | - | Serious adverse effects in 3.8% of patients |
| Bitopertin mild dose | 40.7 ± 9.4 | 81 | 30 mg/day | Improvement in PANSS and NSFS score | Serious adverse effects in 2.4% of patients | |||||
| Bitopertin low dose | 41.1 ± 10.4 | 82 | 10 mg/day | Improvement in PANSS, NSFS, CGI-I-N, and PSP score | Serious adverse effects in 1.21% of patients | |||||
| Placebo | 30.0 ± 10.8 | 81 | - | - | ||||||
| Bugarski-Kirola et al., 2016 [356] | RCT | Bitopertin high dose | 39.1 ± 12.2 | 202 | 20 mg/day | 12 weeks | Unspecified | - | 1% | |
| Bitopertin low dose | 40.2 ± 12.4 | 200 | 10 mg/day | Improvement in PANSS and PSFS score compared to placebo | 2% three deaths due to upper gastrointestinal hemorrhage caused by duodenal ulcer, alcohol poisoning and related head injury, and suicide | |||||
| Placebo | 39.7 ± 12.7 | 200 | - | - | 3.5% | |||||
| Kantrowitz et al., 2017 [357] | RCT | Bitopertin | 40 ± 11 | 17 | 10 mg/day | 6 weeks | CPZ equivalents > 600 mg | No differences between groups | No side effect | |
| Placebo | 43 ± 12 | 12 | - | |||||||
| BI425809 | Fleischhacker et al., 2021 [358] | RCT | BI425809 | 36.5 ± 8.5 | 85 | 2 mg/day | 12 weeks | Unspecified antipsychotics treatment | Improvement in cognitive functions in BI425809 groups compared to placebo. The largest enhancement from baseline vs. placebo was observed in the 10 mg and 25 mg dose group | Headache Somnolence Gastrointestinal disorders |
| BI425809 | 37.5 ± 7.9 | 84 | 5 mg/day | |||||||
| BI425809 | 37.9 ± 6.8 | 85 | 10 mg/day | |||||||
| BI425809 | 36.2 ± 7.8 | 85 | 25 mg/day | |||||||
| Placebo | 37.2 ± 7.7 | 170 | - | |||||||
| D-cycloserine | Cain et al., 2014 [359] | RCT | D-cycloserine | 48.8 ± 11.5 | 18 | 50 mg/day | 8 weeks | Clozapine and others (unspecified) | Improvement in performance on the auditory discrimination task, working memory, and negative symptoms | - |
| Placebo | 46.2 ± 13.3 | 18 | ||||||||
| Duncan et al., 2004 [360] | RCT | D-cycloserine | 48.7 ± 12.1 | 10 | 250 mg/day | 4 weeks | Unspecified | Improvement in SANS and BPRS score | No side effects | |
| Placebo | 54.4 ± 11.8 | 12 | ||||||||
| Goff et al., 1999 [361] | RCT | D-cycloserine | 36.6 ± 9.6 | 17 | 50 mg/day | 6 weeks | Unspecified | Worsening in negative symptoms | - | |
| Placebo | ||||||||||
| Goff et al., 1999 [362] | RCT | D-cycloserine | 46.8 ± 12.3 | 23 | 50 mg/day | 8 weeks | Unspecified | Improvement in SANS score | - | |
| Placebo | 41.2 ± 8.1 | 24 | ||||||||
| Goff et al., 2005 [363] | RCT | D-cycloserine | 45.9 ± 7.4 | 22 | 50 mg/day | 6 months | Unspecified | No significant differences | 9% | |
| Placebo | 47.0 ± 8.6 | 28 | 21% | |||||||
| Goff et al., 2008 [364] | RCT | D-cycloserine | 50.1 ± 9.15 | 19 | 50 mg/day | 8 weeks | All antipsychotic except for clozapine | Improvement in SANS score | No side effects | |
| Placebo | 48.0 ± 6.66 | 19 | ||||||||
| Heresco-Levy et al., 2002 [365] | RCT | D-cycloserine | 40.0 ± 12.1 | 8 | 50 mg/day | 6 weeks | Conventional antipsychotics and olanzapine, or risperidone | Improvement in SANS and PANSS negative score | Worsening of psychotic symptoms in two patients on treatment and two during placebo | |
| Placebo | 8 | |||||||||
| Rosse et al., 1996 [366] | Open-label study | D-cycloserine high dose | 38.1 ± 6.8 | 6 | 30 mg/day | 4 weeks | Molindone 150 mg/day | No significant differences | No side effects | |
| D-cycloserine low dose | 3 | 10 mg/day | ||||||||
| Placebo | 4 | - | ||||||||
| Takiguchi et al., 2017 [367] | RCT crossover study | D-cycloserine | 44.0 ± 14.0 | 36 | 50 mg/day | 6 weeks | Unspecified: 827.0 ± 609.5 CPZ equivalents | No significant differences | Liver enzyme elevation in two patients of the placebo group | |
| Placebo |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bartolomeis, A.; Vellucci, L.; Austin, M.C.; De Simone, G.; Barone, A. Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics. Biomolecules 2022, 12, 909. https://doi.org/10.3390/biom12070909
de Bartolomeis A, Vellucci L, Austin MC, De Simone G, Barone A. Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics. Biomolecules. 2022; 12(7):909. https://doi.org/10.3390/biom12070909
Chicago/Turabian Stylede Bartolomeis, Andrea, Licia Vellucci, Mark C. Austin, Giuseppe De Simone, and Annarita Barone. 2022. "Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics" Biomolecules 12, no. 7: 909. https://doi.org/10.3390/biom12070909
APA Stylede Bartolomeis, A., Vellucci, L., Austin, M. C., De Simone, G., & Barone, A. (2022). Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics. Biomolecules, 12(7), 909. https://doi.org/10.3390/biom12070909