
The Role of Hsp90 in Retinal Proteostasis and Disease


Abstract
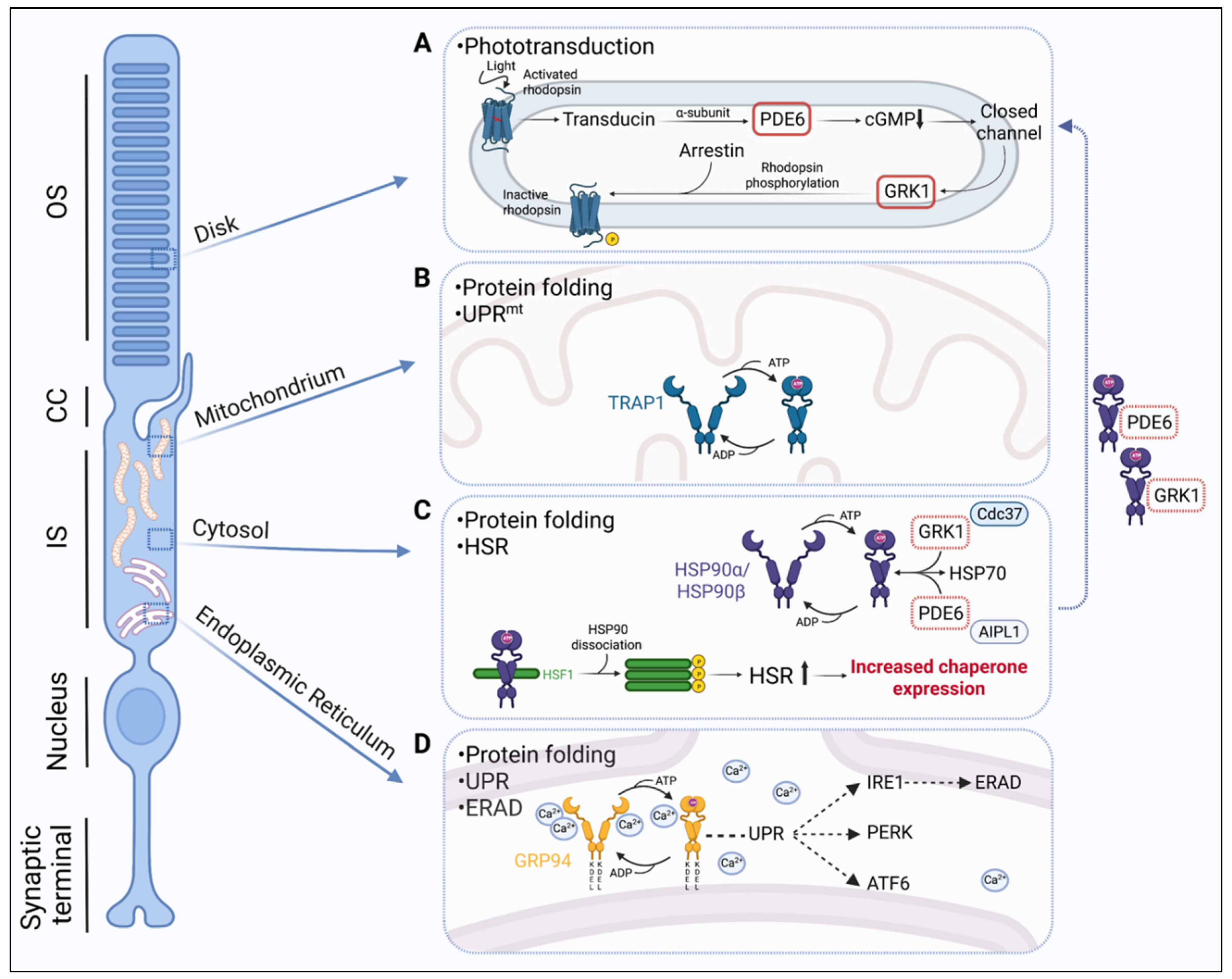
:1. Phototransduction and Protein Folding in Photoreceptors (PR)
2. The Importance of Hsp90 Isoforms in Retinal Proteostasis
2.1. Cytosolic Hsp90
2.2. ER-Associated GRP94
2.3. Mitochondrial TRAP1
3. The Role of Hsp90 in Retinal Disease
3.1. Hsp90 and the Stress Response in Retinal Disease
3.2. Hsp90 Inhibition in Retinal Disease
3.3. Ocular Toxicities in Clinical Trials of Hsp90 Inhibition
4. Hsp90 Client Proteins in the Retina
4.1. The Hsp90-PDE6 Chaperone Complex
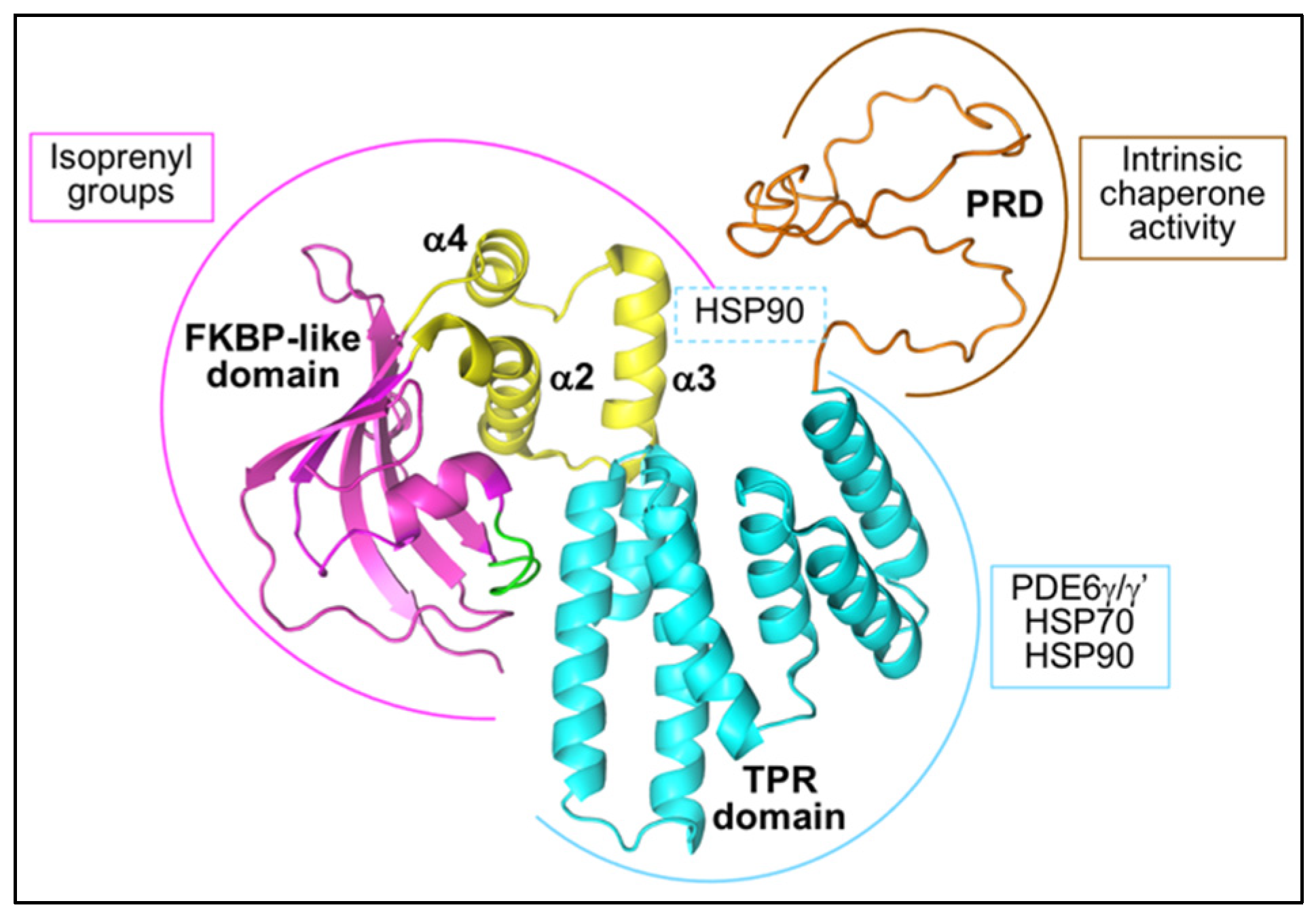
4.1.1. AIPL1 Structure
4.1.2. The Interaction of AIPL1 with Hsp90
4.1.3. The AIPL1-Mediated Targeting of PDE6 to Hsp90
4.2. The Hsp90-GRK1 Chaperone Complex
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshavsky, V.Y.; Lamb, T.D.; Pugh, E.N. G Proteins and Phototransduction. Annu. Rev. Physiol. 2003, 64, 153–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshavsky, V.Y.; Wensel, T.G. Timing Is Everything: GTPase Regulation in Phototransduction. Investig. Opthalmology Vis. Sci. 2013, 54, 7725–7733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefalov, V. Phototransduction: Phototransduction in Cones. Encycl. Eye 2010, 389–396. [Google Scholar] [CrossRef]
- Ingram, N.T.; Sampath, A.P.; Fain, G.L. Why are rods more sensitive than cones? J. Physiol. 2016, 594, 5415–5426. [Google Scholar] [CrossRef] [Green Version]
- Spencer, W.J.; Lewis, T.; Pearring, J.N.; Arshavsky, V.Y. Photoreceptor Discs: Built Like Ectosomes. Trends Cell Biol. 2020, 30, 904–915. [Google Scholar] [CrossRef]
- Léveillard, T.; Sahel, J.-A. Metabolic and redox signaling in the retina. In Cellular and Molecular Life Sciences; Birkhauser Verlag AG: Basel, Switzerland, 2017; Volume 74, Issue 20; pp. 3649–3665. [Google Scholar] [CrossRef] [Green Version]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.M.; Casson, R.J. Glucose metabolism in mammalian photoreceptor inner and outer segments. Clin. Exp. Ophthalmol. 2017, 45, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef] [PubMed]
- Åkerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Voellmy, R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperon. 2004, 9, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Luengo, T.M.; Kityk, R.; Mayer, M.P.; Rüdiger, S.G. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell 2018, 70, 545–552.e9. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zheng, X.; Ding, Y.; Zhou, M.; Wei, Z.; Liu, T.; Liao, K. The molecular chaperone Hsp90α deficiency causes retinal degeneration by disrupting Golgi organization and vesicle transportation in photoreceptors. J. Mol. Cell Biol. 2020, 12, 216–229. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. et Biophys. Acta-Mol. Cell Res. 2012, 1823, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Meunier, L.; Usherwood, Y.-K.; Chung, K.T.; Hendershot, L.M. A Subset of Chaperones and Folding Enzymes Form Multiprotein Complexes in Endoplasmic Reticulum to Bind Nascent Proteins. Mol. Biol. Cell 2002, 13, 4456–4469. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Anukanth, A.; Khorana, H. Structure and function in rhodopsin. Requirements of a specific structure for the intradiscal domain. J. Biol. Chem. 1994, 269, 19738–19744. [Google Scholar] [CrossRef]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroeger, H.; Messah, C.; Ahern, K.; Gee, J.; Joseph, V.; Matthes, M.T.; Yasumura, D.; Gorbatyuk, M.S.; Chiang, W.-C.; LaVail, M.M.; et al. Induction of Endoplasmic Reticulum Stress Genes, BiP and Chop, in Genetic and Environmental Models of Retinal Degeneration. Investig. Opthalmology Vis. Sci. 2012, 53, 7590–7599. [Google Scholar] [CrossRef] [Green Version]
- Saliba, R.S.; Munro, P.M.G.; Luthert, P.J.; Cheetham, M.E. The cellular fate of mutant rhodopsin: Quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2017, 70, 24–31. [Google Scholar] [CrossRef]
- Di Jeso, B.; Ulianich, L.; Pacifico, F.; Leonardi, A.; Vito, P.; Consiglio, E.; Formisano, S.; Arvan, P. Folding of thyroglobulin in the calnexin/calreticulin pathway and its alteration by loss of Ca2+ from the endoplasmic reticulum. Biochem. J. 2003, 370, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Preissler, S.; Rato, C.; Yan, Y.; Perera, L.A.; Czako, A.; Ron, D. Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising BiP-substrate complexes. eLife 2020, 9, 1–36. [Google Scholar] [CrossRef]
- Wengert, L.A.; Backe, S.J.; Bourboulia, D.; Mollapour, M.; Woodford, M.R. TRAP1 Chaperones the Metabolic Switch in Cancer. Biomolecules 2022, 12, 786. [Google Scholar] [CrossRef]
- Felts, S.J.; Owen, B.A.L.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related Protein TRAP1 Is a Mitochondrial Protein with Distinct Functional Properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [Green Version]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.W.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Baqri, R.M.; Pietron, A.V.; Gokhale, R.H.; Turner, B.A.; Kaguni, L.S.; Shingleton, A.W.; Kunes, S.; Miller, K.E. Mitochondrial chaperone TRAP1 activates the mitochondrial UPR and extends healthspan in Drosophila. Mech. Ageing Dev. 2014, 141–142, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, K.; Miyata, S.; Takamura, H.; Katayama, T.; Tohyama, M. Mitochondrial TRAP1 regulates the unfolded protein response in the endoplasmic reticulum. Neurochem. Int. 2011, 58, 880–887. [Google Scholar] [CrossRef]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Inherited retinal diseases: Therapeutics, clinical trials and end points—A review. Clin. Exp. Ophthalmol. 2021, 49, 270–288. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef] [Green Version]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilà, M.; Bevilacqua, D.; McCulley, C.; Schwarz, N.; Athanasiou, D.; Kanuga, N.; Novoselov, S.S.; Lange, C.A.; Ali, R.R.; Bainbridge, J.W.; et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum. Mol. Genet. 2014, 23, 2164–2175. [Google Scholar] [CrossRef] [Green Version]
- Kennan, A.; Aherne, A.; Palfi, A.; Humphries, M.; McKee, A.; Stitt, A.; Simpson, D.A.C.; Demtroder, K.; Orntoft, T.; Ayuso, C.; et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho-/- mice. Hum. Mol. Genet. 2002, 11, 547–558. [Google Scholar] [CrossRef]
- Tam, L.C.; Kiang, A.-S.; Campbell, M.; Keaney, J.; Farrar, G.J.; Humphries, M.M.; Kenna, P.F.; Humphries, P. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90). Hum. Mol. Genet. 2010, 19, 4421–4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Liu, Y.; Ye, J.; Ying, W.; Ogawa, L.S.; Inoue, T.; Tatsuta, N.; Wada, Y.; Koya, K.; Huang, Q.; et al. A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol. Appl. Pharmacol. 2013, 273, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummar, S.; Gutierrez, M.E.; Gardner, E.R.; Chen, X.; Figg, W.D.; Zajac-Kaye, M.; Chen, M.; Steinberg, S.M.; Muir, C.A.; Yancey, M.A.; et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Samuel, T.A.; Sessa, C.; Britten, C.; Milligan, K.S.; Mita, M.M.; Banerji, U.; Pluard, T.J.; Stiegler, P.; Quadt, C.; Shapiro, G. AUY922, a novel HSP90 inhibitor: Final results of a first-in-human study in patients with advanced solid malignancies. J. Clin. Oncol. 2010, 28, 2528. [Google Scholar] [CrossRef]
- Shapiro, G.; Kwak, E.L.; Dezube, B.J.; Lawrence, D.P.; Cleary, J.M.; Lewis, S.; Squires, M.; Lock, V.; Lyons, J.F.; Yule, M. Phase I pharmacokinetic and pharmacodynamic study of the heat shock protein 90 inhibitor AT13387 in patients with refractory solid tumors. J. Clin. Oncol. 2010, 28, 3069. [Google Scholar] [CrossRef]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.-T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A Phase I Study of the Heat Shock Protein 90 Inhibitor Alvespimycin (17-DMAG) Given Intravenously to Patients with Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Rajan, A.; Kelly, R.J.; Trepel, J.B.; Kim, Y.S.; Alarcon, S.V.; Kummar, S.; Gutierrez, M.; Crandon, S.; Zein, W.M.; Jain, L.; et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin. Cancer Res. 2011, 17, 6831–6839. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Jones, S.F.; Hart, L.; Pant, S.; Moyhuddin, A.; Lane, C.M.; Earwood, C.; Murphy, P.; Patton, J.; Penley, W.C.; et al. A Phase I Study of the Hsp90 Inhibitor AUY922 plus Capecitabine for the Treatment of Patients with Advanced Solid Tumors. Cancer Investig. 2015, 33, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Seggewiss-Bernhardt, R.; Bargou, R.C.; Goh, Y.T.; Stewart, A.K.; Spencer, A.; Alegre, A.; Bladé, J.; Ottmann, O.G.; Fernandez-Ibarra, C.; Lu, H.; et al. Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer 2015, 121, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Bauer, T.M.; Lamar, R.; Joseph, M.; Penley, W.; Thompson, D.S.; Spigel, D.R.; Owera, R.; Lane, C.M.; Earwood, C.; et al. A Phase 2 Study of the Hsp90 Inhibitor AUY922 as Treatment for Patients with Refractory Gastrointestinal Stromal Tumors. Cancer Investig. 2016, 34, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, N.-J.; Yeh, K.-H.; Chiu, C.-F.; Chen, J.-S.; Yen, C.-C.; Lee, K.-D.; Lin, Y.-L.; Bai, L.-Y.; Chen, M.-H.; Lin, J.-S.; et al. Results of Phase II trial of AUY922, a novel heat shock protein inhibitor in patients with metastatic gastrointestinal stromal tumor (GIST) and imatinib and sunitinib therapy. J. Clin. Oncol. 2016, 34, 134. [Google Scholar] [CrossRef]
- Do, K.T.; Coyne, G.O.; Hays, J.L.; Supko, J.G.; Liu, S.V.; Beebe, K.; Neckers, L.; Trepel, J.B.; Lee, M.-J.; Smyth, T.; et al. Phase 1 study of the HSP90 inhibitor onalespib in combination with AT7519, a pan-CDK inhibitor, in patients with advanced solid tumors HHS Public Access. Cancer Chemother. Pharmacol. 2020, 86, 815–827. [Google Scholar] [CrossRef]
- Shen, C.H.; Hsieh, C.C.; Jiang, K.Y.; Lin, C.Y.; Chiang, N.J.; Li, T.W.; Yen, C.T.; Chen, W.J.; Hwang, D.Y.; Chen, L.T. AUY922 induces retinal toxicity through attenuating TRPM1. J. Biomed. Sci. 2021, 28, 1–21. [Google Scholar] [CrossRef]
- Kanamaru, C.; Yamada, Y.; Hayashi, S.; Matsushita, T.; Suda, A.; Nagayasu, M.; Kimura, K.; Chiba, S. Retinal toxicity induced by small-molecule Hsp90 inhibitors in beagle dogs. J. Toxicol. Sci. 2014, 39, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Roman, D.; VerHoeve, J.; Schadt, H.; Vicart, A.; Walker, U.J.; Turner, O.; Richardson, T.A.; Wolford, S.T.; Miller, P.E.; Zhou, W.; et al. Ocular toxicity of AUY922 in pigmented and albino rats. Toxicol. Appl. Pharmacol. 2016, 309, 55–62. [Google Scholar] [CrossRef]
- Cote, R.H. Photoreceptor phosphodiesterase (PDE6): Activation and inactivation mechanisms during visual transduction in rods and cones. Pflug. Arch. Eur. J. Physiol. 2021, 473, 1377–1391. [Google Scholar] [CrossRef]
- Sohocki, M.M.; Bowne, S.J.; Sullivan, L.S.; Blackshaw, S.; Cepko, C.L.; Payne, A.; Bhattacharya, S.S.; Khaliq, S.; Mehdi, S.Q.; Birch, D.; et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet. 2000, 24, 79–83. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Wen, X.H.; Woodruff, M.L.; Pawlyk, B.; Yang, J.; Fain, G.L.; Sandberg, M.A.; Makino, C.L.; Li, T. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13903–13908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamurthy, V.; Niemi, G.A.; Reh, T.A.; Hurley, J.B. Leber congenital amaurosis linked to AIPL1: A mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13897–13902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschman, L.T.; Kolandaivelu, S.; Frederick, J.M.; Dang, L.; Goldberg, A.F.; Baehr, W.; Ramamurthy, V. The Leber congenital amaurosis protein, AIPL1, is needed for the viability and functioning of cone photoreceptor cells. Hum. Mol. Genet. 2009, 19, 1076–1087. [Google Scholar] [CrossRef]
- Kolandaivelu, S.; Huang, J.; Hurley, J.B.; Ramamurthy, V. AIPL1, a Protein Associated with Childhood Blindness, Interacts with α-Subunit of Rod Phosphodiesterase (PDE6) and Is Essential for Its Proper Assembly. J. Biol. Chem. 2009, 284, 30853–30861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolandaivelu, S.; Singh, R.K.; Ramamurthy, V. AIPL1, A protein linked to blindness, is essential for the stability of enzymes mediating cGMP metabolism in cone photoreceptor cells. Hum. Mol. Genet. 2013, 23, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.P.; Boyd, K.; Yu, L.; Artemyev, N.O. Interaction of the tetratricopeptide repeat domain of aryl hydrocarbon receptor–interacting protein–like 1 with the regulatory Pγ subunit of phosphodiesterase 6. J. Biol. Chem. 2019, 294, 15795–15807. [Google Scholar] [CrossRef]
- Li, J.; Zoldak, G.; Kriehuber, T.; Soroka, J.; Schmid, F.X.; Richter, K.; Buchner, J. Unique Proline-Rich Domain Regulates the Chaperone Function of AIPL1. Biochemistry 2013, 52, 2089–2096. [Google Scholar] [CrossRef]
- Linnert, M.; Lin, Y.-J.; Manns, A.; Haupt, K.; Paschke, A.-K.; Fischer, G.; Weiwad, M.; Lücke, C. The FKBP-Type Domain of the Human Aryl Hydrocarbon Receptor-Interacting Protein Reveals an Unusual Hsp90 Interaction. Biochemistry 2013, 52, 2097–2107. [Google Scholar] [CrossRef]
- Majumder, A.; Gopalakrishna, K.N.; Cheguru, P.; Gakhar, L.; Artemyev, N.O. Interaction of Aryl Hydrocarbon Receptor-interacting Protein-like 1 with the Farnesyl Moiety. J. Biol. Chem. 2013, 288, 21320–21328. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.P.; Gakhar, L.; Yu, L.; Artemyev, N.O. Unique structural features of the AIPL1–FKBP domain that support prenyl lipid binding and underlie protein malfunction in blindness. Proc. Natl. Acad. Sci. USA 2017, 114, E6536–E6545. [Google Scholar] [CrossRef] [Green Version]
- Sohocki, M.M.; Sullivan, L.S.; Tirpak, D.L.; Daiger, S.P. Comparative analysis of aryl-hydrocarbon receptor interacting protein-like 1 (Aipl1), a gene associated with inherited retinal disease in humans. Mamm. Genome 2001, 12, 566–568. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.P.; Majumder, A.; Gakhar, L.; Artemyev, N.O. Extended conformation of the proline-rich domain of human aryl hydrocarbon receptor-interacting protein-like 1: Implications for retina disease. J. Neurochem. 2015, 135, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-De-Quintana, J.; Evans, R.J.; Cheetham, M.; Van Der Spuy, J. The Leber Congenital Amaurosis Protein AIPL1 Functions as Part of a Chaperone Heterocomplex. Investig. Opthalmology Vis. Sci. 2008, 49, 2878–2887. [Google Scholar] [CrossRef] [Green Version]
- Sacristan-Reviriego, A.; Bellingham, J.; Prodromou, C.; Boehm, A.N.; Aichem, A.; Kumaran, N.; Bainbridge, J.; Michaelides, M.; Van Der Spuy, J. The integrity and organization of the human AIPL1 functional domains is critical for its role as a HSP90-dependent co-chaperone for rod PDE6. Hum. Mol. Genet. 2017, 26, 4465–4480. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.P.; Boyd, K.; Artemyev, N.O. Molecular insights into the maturation of phosphodiesterase 6 by the specialized chaperone complex of HSP90 with AIPL1. J. Biol. Chem. 2022, 298, 101620. [Google Scholar] [CrossRef]
- Sacristan-Reviriego, A.; Le, H.M.; Georgiou, M.; Meunier, I.; Bocquet, B.; Roux, A.-F.; Prodromou, C.; Bainbridge, J.; Michaelides, M.; van der Spuy, J. Clinical and functional analyses of AIPL1 variants reveal mechanisms of pathogenicity linked to different forms of retinal degeneration. Sci. Rep. 2020, 10, 17520. [Google Scholar] [CrossRef]
- Cheung-Flynn, J.; Roberts, P.J.; Riggs, D.L.; Smith, D.F. C-terminal Sequences outside the Tetratricopeptide Repeat Domain of FKBP51 and FKBP52 Cause Differential Binding to Hsp90. J. Biol. Chem. 2003, 278, 17388–17394. [Google Scholar] [CrossRef] [Green Version]
- van der Spuy, J.; Cheetham, M. The Leber Congenital Amaurosis Protein AIPL1 Modulates the Nuclear Translocation of NUB1 and Suppresses Inclusion Formation by NUB1 Fragments. J. Biol. Chem. 2004, 279, 48038–48047. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Yadav, R.P.; Artemyev, N.O. NMR resonance assignments of the FKBP domain of human aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) in complex with a farnesyl ligand. Biomol. NMR Assign. 2017, 11, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamurthy, V.; Roberts, M.; Van den Akker, F.; Niemi, G.; Reh, T.A.; Hurley, J.B. AIPL1, a protein implicated in Leber’s congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12630–12635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishna, K.N.; Boyd, K.; Yadav, R.P.; Artemyev, N.O. Aryl Hydrocarbon Receptor-interacting Protein-like 1 Is an Obligate Chaperone of Phosphodiesterase 6 and Is Assisted by the γ-Subunit of Its Client. J. Biol. Chem. 2016, 291, 16282–16291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, J.; Neuhauss, S.C.F. The Binding Properties and Physiological Functions of Recoverin. Front. Mol. Neurosci. 2018, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Plasencia, M.; Li, Z.; Mukherjee, S.; Patra, D.; Chen, C.-L.; Klose, T.; Yao, X.-Q.; Kossiakoff, A.A.; Chang, L.; et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 2021, 595, 600–605. [Google Scholar] [CrossRef]
- Yeung, W.; Ruan, Z.; Kannan, N. Emerging roles of the αC-β4 loop in protein kinase structure, function, evolution, and disease. IUBMB Life 2020, 72, 1189–1202. [Google Scholar] [CrossRef]
- Luo, J.; Benovic, J.L. G Protein-coupled Receptor Kinase Interaction with Hsp90 Mediates Kinase Maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yuan, X.; Xiang, Z.; Mimnaugh, E.; Marcu, M.; Neckers, L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 2005, 12, 120–126. [Google Scholar] [CrossRef]
- Citri, A.; Harari, D.; Shohat, G.; Ramakrishnan, P.; Gan, J.; Lavi, S.; Eisenstein, M.; Kimchi, A.; Wallach, D.; Pietrokovski, S.; et al. Hsp90 Recognizes a Common Surface on Client Kinases. J. Biol. Chem. 2006, 281, 14361–14369. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.J.; Mandal, A.K.; Theodoraki, M. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007, 17, 87–92. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verba, K.A.; Agard, D.A. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 2017, 42, 799–811. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Study Outcome | Reference |
|---|---|---|
| Geldanamycin | Reduced P23H aggregation and cell death in vitro | Mendes & Cheetham, 2008 [42] |
| Tanepsimycin 17-AAG | Reduced P23H aggregation and cell death in vitro | Mendes & Cheetham, 2008 [42] |
| Reduced protein accumulation in R135L rats | Aguilà et al., 2014 [43] | |
| Radicicol | Reduced P23H aggregation and cell death in vitro | Mendes & Cheetham, 2008 [42] |
| Alvespimycin 17-DMAG | Prolonged treatment causes photoreceptor cell death in rats | Zhou et al., 2013 [46] |
| Induced photoreceptor apoptosis and rhodopsin retention in the IS in wild-type mice | Wu et al., 2020 [19] | |
| HSP990 | Reduces P23H aggregation, improves visual function and delays photoreceptor cell death in P23H-1 rats | Aguilà et al., 2014 [43] |
| Protects photoreceptors from degeneration caused by aggregating mutant IMPDH1 protein | Tam et al., 2010 [45] |
| HSP90 Inhibitor Drug | Trial | ClinicalTrials.gov Identifier | Ocular Effect | Reference |
|---|---|---|---|---|
| Alvespimycin 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) | Phase I trial of 17-DMAG in patients with advanced malignancies | NCT00088868 |
| Kummar et al., 2010 [47] |
| Phase I trial of 17-DMAG in patients with advanced solid tumors | NCT00248521 |
| Pacey et al., 2011 [50] | |
| Onalespid AT13387 | Phase I trial of AT13387 in patients with refractory solid tumors. | NCT00878423 |
| Shapiro et al., 2010 [49] |
| Phase I study of onalespib in combination with AT7519, a pan-CDK inhibitor, in patients with advanced solid tumors | NCT02503709 |
| Do et al., 2020 [56] | |
| Luminespid AUY922 NVP-AUY922 | Phase I trial of AUY922 in combination with capecitabine in patients with advanced solid tumors | NCT01226732 |
| Bendell et al., 2015 [52] |
| Phase I-IB/II trial of NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma | NCT00708292 |
| Seggewiss-Bernhardt et al., 2015 [53] | |
| Phase II trial of AUY922 in patients with refractory gastrointestinal stromal tumors | NCT01404650 |
| Bendell et al., 2016 [54] | |
| Phase II trial of AUY922 in patients with metastatic gastrointestinal stromal tumor | NCT01389583 |
| Chiang et al., 2016 [55] Shen et al., 2021 [57] | |
| SNX-5422 PF-04929113 | Phase I study of SNX-5422 in patients with refractory solid tumor malignancies and lymphomas | NCT00644072 |
| Rajan et al., 2011 [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziaka, K.; van der Spuy, J. The Role of Hsp90 in Retinal Proteostasis and Disease. Biomolecules 2022, 12, 978. https://doi.org/10.3390/biom12070978
Ziaka K, van der Spuy J. The Role of Hsp90 in Retinal Proteostasis and Disease. Biomolecules. 2022; 12(7):978. https://doi.org/10.3390/biom12070978
Chicago/Turabian StyleZiaka, Kalliopi, and Jacqueline van der Spuy. 2022. "The Role of Hsp90 in Retinal Proteostasis and Disease" Biomolecules 12, no. 7: 978. https://doi.org/10.3390/biom12070978
APA StyleZiaka, K., & van der Spuy, J. (2022). The Role of Hsp90 in Retinal Proteostasis and Disease. Biomolecules, 12(7), 978. https://doi.org/10.3390/biom12070978