
Emerging Link between Tsc1 and FNIP Co-Chaperones of Hsp90 and Cancer

 , and
, and
Abstract
:1. Introduction
2. FNIP1 and FNIP2
2.1. FNIP1/2 Structure and Function
2.2. FNIP1 Function in Skeletal Muscle and Adipocytes
2.3. FNIP1 in Oxidative Stress
2.4. FNIP1 Function in B-Cell Development
2.5. Role of FNIP1/2 in Transcription
3. Tsc1
3.1. Structure and Function of the Tsc1/2 Complex
3.2. Separable Functions of Tsc1 and Tsc2
3.3. mTOR Independent Functions of Tsc1
4. Regulation of Hsp90 Chaperone Function by Co-Chaperones
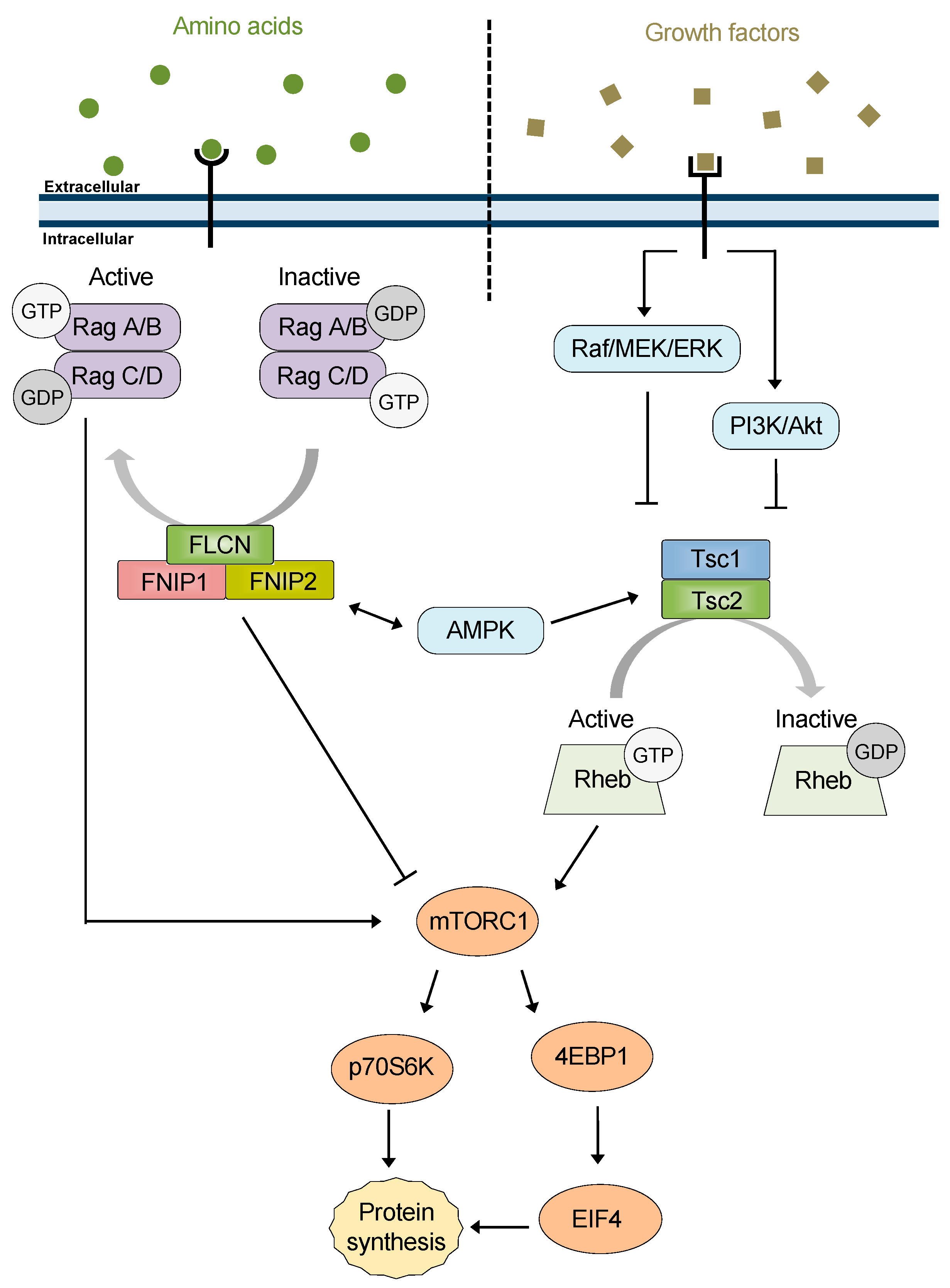
5. FNIP1/2 and Tsc1: New Co-Chaperones of Hsp90
5.1. FNIP1/2 and Tsc1 in the Chaperone Cycle
5.2. FNIPs, Tsc1 and the Chaperone Code
6. A New Perspective: FNIPs, Tsc1, and mTOR
6.1. FNIP1/2 Co-Chaperone Activity Contributes to mTOR Regulation
6.2. Co-Chaperone Activity of Tsc1 in Regulation of mTOR
7. Specialized Function of FNIP1/2 and Tsc1: Chaperoning Tumor Suppressors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Wegele, H.; Muschler, P.; Bunck, M.; Reinstein, J.; Buchner, J. Dissection of the contribution of individual domains to the ATPase mechanism of Hsp90. J. Biol. Chem. 2003, 278, 39303–39310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromou, C. The ‘active life’ of Hsp90 complexes. Biochim. Biophys. Acta 2012, 1823, 614–623. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Beebe, K.; Neckers, L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 2009, 5, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol. Cell 2019, 74, 831–843. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Singer, M.A.; Lindquist, S. Maturation of the tyrosine kinase c-src as a kinase and as a substrate depends on the molecular chaperone Hsp90. Proc. Natl. Acad. Sci. USA 1999, 96, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckers, L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol. Med. 2002, 8, S55–S61. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update from 2014. Curr. Drug Targets 2020, 21, 302–317. [Google Scholar] [CrossRef]
- Cox, M.B.; Johnson, J.L. The Role of p23, Hop, Immunophilins, and Other Co-chaperones in Regulating Hsp90 Function. Methods Mol. Biol. 2011, 787, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Ratzke, C.; Hellenkamp, B.; Hugel, T. Four-colour FRET reveals directionality in the Hsp90 multicomponent machinery. Nat. Commun. 2014, 5, 4192. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The Plasticity of the Hsp90 Co-chaperone System. Mol. Cell 2017, 67, 947–961.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, V.; Rutz, D.A.; Moessmer, P.; Muhlhofer, M.; Lawatscheck, J.; Rief, M.; Buchner, J. The switch from client holding to folding in the Hsp70/Hsp90 chaperone machineries is regulated by a direct interplay between co-chaperones. Mol. Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Hong, S.B.; Sharma, N.; Warren, M.B.; Nickerson, M.L.; Iwamatsu, A.; Esposito, D.; Gillette, W.K.; Hopkins, R.F., 3rd; Hartley, J.L.; et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15552–15557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasumi, H.; Baba, M.; Hong, S.B.; Hasumi, Y.; Huang, Y.; Yao, M.; Valera, V.A.; Linehan, W.M.; Schmidt, L.S. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 2008, 415, 60–67. [Google Scholar] [CrossRef] [Green Version]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Dunn, D.M.; Blanden, A.R.; Capriotti, D.; Loiselle, D.; Prodromou, C.; Panaretou, B.; Hughes, P.F.; Smith, A.; Ackerman, W.; et al. The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat. Commun. 2016, 7, 12037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodford, M.R.; Sager, R.A.; Marris, E.; Dunn, D.M.; Blanden, A.R.; Murphy, R.L.; Rensing, N.; Shapiro, O.; Panaretou, B.; Prodromou, C.; et al. Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 2017, 36, 3650–3665. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat. Rev. Urol. 2015, 12, 558–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sager, R.A.; Woodford, M.R.; Shapiro, O.; Mollapour, M.; Bratslavsky, G. Sporadic renal angiomyolipoma in a patient with Birt-Hogg-Dube: Chaperones in pathogenesis. Oncotarget 2018, 9, 22220–22229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, L.; Stein, A.; Gronbaek-Thygesen, M.; Nygaard, L.; Soltoft, C.L.; Nielsen, S.V.; Lisby, M.; Ravid, T.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Folliculin variants linked to Birt-Hogg-Dube syndrome are targeted for proteasomal degradation. PLoS Genet. 2020, 16, e1009187. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Rogala, K.B.; Chou, H.T.; Huang, R.K.; Yu, Z.; Sabatini, D.M. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 2019, 179, 1319–1329.e8. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Ferguson, S.M. GATOR1-dependent recruitment of FLCN-FNIP to lysosomes coordinates Rag GTPase heterodimer nucleotide status in response to amino acids. J. Cell Biol. 2018, 217, 2765–2776. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc. Nutr. Soc. 2011, 70, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur, S.D.; et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Liu, J.; Sun, Z.; Yin, Y.; Mao, Y.; Xu, D.; Liu, L.; Xu, Z.; Guo, Q.; Ding, C.; et al. AMPK-dependent and -independent coordination of mitochondrial function and muscle fiber type by FNIP1. PLoS Genet. 2021, 17, e1009488. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, X.; Zhou, D.; Lai, L.; Xiao, L.; Liu, L.; Fu, T.; Kong, Y.; Zhou, Q.; Vega, R.B.; et al. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. EMBO Mol. Med. 2016, 8, 1212–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Wang, H.; Liao, Y.; Zhou, P.; Xu, Y.; Zhao, Y.; Xie, S.; Zhao, S.; Li, X. miR-208b modulating skeletal muscle development and energy homoeostasis through targeting distinct targets. RNA Biol. 2020, 17, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gu, Y.; Lang, H.; Wang, X.; Chen, K.; Gong, X.; Zhou, M.; Ran, L.; Zhu, J.; Mi, M. Dihydromyricetin prevents obesity-induced slow-twitch-fiber reduction partially via FLCN/FNIP1/AMPK pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1282–1291. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, D.; Mao, Y.; Xiao, L.; Sun, Z.; Liu, J.; Zhou, D.; Xu, Z.; Liu, L.; Fu, T.; et al. FNIP1 regulates adipocyte browning and systemic glucose homeostasis in mice by shaping intracellular calcium dynamics. J. Exp. Med. 2022, 219, e20212491. [Google Scholar] [CrossRef]
- Manford, A.G.; Rodriguez-Perez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61. [Google Scholar] [CrossRef]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martinez-Gonzalez, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodriguez-Perez, F.; et al. Structural basis and regulation of the reductive stress response. Cell 2021, 184, 5375–5390.e16. [Google Scholar] [CrossRef]
- Cai, W.; Yang, H. The structure and regulation of Cullin 2 based E3 ubiquitin ligases and their biological functions. Cell Div. 2016, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Woodford, M.R.; Baker-Williams, A.J.; Sager, R.A.; Backe, S.J.; Blanden, A.R.; Hashmi, F.; Kancherla, P.; Gori, A.; Loiselle, D.R.; Castelli, M.; et al. The tumor suppressor folliculin inhibits lactate dehydrogenase A and regulates the Warburg effect. Nat. Struct. Mol. Biol. 2021, 28, 662–670. [Google Scholar] [CrossRef]
- Park, H.; Staehling, K.; Tsang, M.; Appleby, M.W.; Brunkow, M.E.; Margineantu, D.; Hockenbery, D.M.; Habib, T.; Liggitt, H.D.; Carlson, G.; et al. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity 2012, 36, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.A.; Iwata, T.; Park, H.; Tsang, M.; Kang, J.; Cui, K.; Kwong, W.; James, R.G.; Baba, M.; Schmidt, L.S.; et al. Folliculin Interacting Protein 1 Maintains Metabolic Homeostasis during B Cell Development by Modulating AMPK, mTORC1, and TFE3. J. Immunol. 2019, 203, 2899–2908. [Google Scholar] [CrossRef]
- Park, H.; Tsang, M.; Iritani, B.M.; Bevan, M.J. Metabolic regulator Fnip1 is crucial for iNKT lymphocyte development. Proc. Natl. Acad. Sci. USA 2014, 111, 7066–7071. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Keller, J.R.; Sun, H.W.; Resch, W.; Kuchen, S.; Suh, H.C.; Hasumi, H.; Hasumi, Y.; Kieffer-Kwon, K.R.; Gonzalez, C.G.; et al. The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dube syndrome is required for murine B-cell development. Blood 2012, 120, 1254–1261. [Google Scholar] [CrossRef]
- Niehues, T.; Ozgur, T.T.; Bickes, M.; Waldmann, R.; Schoning, J.; Brasen, J.; Hagel, C.; Ballmaier, M.; Klusmann, J.H.; Niedermayer, A.; et al. Mutations of the gene FNIP1 associated with a syndromic autosomal recessive immunodeficiency with cardiomyopathy and pre-excitation syndrome. Eur. J. Immunol. 2020, 50, 1078–1080. [Google Scholar] [CrossRef]
- Saettini, F.; Poli, C.; Vengoechea, J.; Bonanomi, S.; Orellana, J.C.; Fazio, G.; Rodriguez, F.H.; Noguera, L.P.; Booth, C.; Jarur-Chamy, V.; et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin-interacting protein 1 deficiency. Blood 2021, 137, 493–499. [Google Scholar] [CrossRef]
- Siggs, O.M.; Stockenhuber, A.; Deobagkar-Lele, M.; Bull, K.R.; Crockford, T.L.; Kingston, B.L.; Crawford, G.; Anzilotti, C.; Steeples, V.; Ghaffari, S.; et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. USA 2016, 113, E3706–E3715. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Audet-Walsh, E.; Manteghi, S.; Dufour, C.R.; Walker, B.; Baba, M.; St-Pierre, J.; Giguere, V.; Pause, A. Chronic AMPK activation via loss of FLCN induces functional beige adipose tissue through PGC-1alpha/ERRalpha. Genes Dev. 2016, 30, 1034–1046. [Google Scholar] [CrossRef] [Green Version]
- El-Houjeiri, L.; Possik, E.; Vijayaraghavan, T.; Paquette, M.; Martina, J.A.; Kazan, J.M.; Ma, E.H.; Jones, R.; Blanchette, P.; Puertollano, R.; et al. The Transcription Factors TFEB and TFE3 Link the FLCN-AMPK Signaling Axis to Innate Immune Response and Pathogen Resistance. Cell Rep. 2019, 26, 3613–3628.e3616. [Google Scholar] [CrossRef] [Green Version]
- El-Houjeiri, L.; Biondini, M.; Paquette, M.; Kuasne, H.; Pacis, A.; Park, M.; Siegel, P.M.; Pause, A. Folliculin impairs breast tumor growth by repressing TFE3-dependent induction of the Warburg effect and angiogenesis. J. Clin. Investig. 2021, 131, e144871. [Google Scholar] [CrossRef]
- Endoh, M.; Baba, M.; Endoh, T.; Hirayama, A.; Nakamura-Ishizu, A.; Umemoto, T.; Hashimoto, M.; Nagashima, K.; Soga, T.; Lang, M.; et al. A FLCN-TFE3 Feedback Loop Prevents Excessive Glycogenesis and Phagocyte Activation by Regulating Lysosome Activity. Cell Rep. 2020, 30, 1823–1834.e1825. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.B.; Oh, H.; Valera, V.A.; Baba, M.; Schmidt, L.S.; Linehan, W.M. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS ONE 2010, 5, e15793. [Google Scholar] [CrossRef] [Green Version]
- Glykofridis, I.E.; Knol, J.C.; Balk, J.A.; Westland, D.; Pham, T.V.; Piersma, S.R.; Lougheed, S.M.; Derakhshan, S.; Veen, P.; Rooimans, M.A.; et al. Loss of FLCN-FNIP1/2 induces a non-canonical interferon response in human renal tubular epithelial cells. eLife 2021, 10, e61630. [Google Scholar] [CrossRef]
- Hasumi, H.; Baba, M.; Hasumi, Y.; Lang, M.; Huang, Y.; Oh, H.F.; Matsuo, M.; Merino, M.J.; Yao, M.; Ito, Y.; et al. Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc. Natl. Acad. Sci. USA 2015, 112, E1624–E1631. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Furihata, M.; Hong, S.B.; Tessarollo, L.; Haines, D.C.; Southon, E.; Patel, V.; Igarashi, P.; Alvord, W.G.; Leighty, R.; et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 2008, 100, 140–154. [Google Scholar] [CrossRef] [Green Version]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef]
- Woodford, M.R.; Backe, S.J.; Sager, R.A.; Bourboulia, D.; Bratslavsky, G.; Mollapour, M. The Role of Heat Shock Protein-90 in the Pathogenesis of Birt-Hogg-Dube and Tuberous Sclerosis Complex Syndromes. Urol. Oncol. 2021, 39, 322–326. [Google Scholar] [CrossRef]
- European Chromosome 16 Tuberous Sclerosis, C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Ramlaul, K.; Fu, W.; Li, H.; de Martin Garrido, N.; He, L.; Trivedi, M.; Cui, W.; Aylett, C.H.S.; Wu, G. Architecture of the Tuberous Sclerosis Protein Complex. J. Mol. Biol. 2021, 433, 166743. [Google Scholar] [CrossRef]
- Fitzian, K.; Bruckner, A.; Brohee, L.; Zech, R.; Antoni, C.; Kiontke, S.; Gasper, R.; Linard Matos, A.L.; Beel, S.; Wilhelm, S.; et al. TSC1 binding to lysosomal PIPs is required for TSC complex translocation and mTORC1 regulation. Mol. Cell 2021, 81, 2705–2721.e8. [Google Scholar] [CrossRef]
- Yang, H.; Yu, Z.; Chen, X.; Li, J.; Li, N.; Cheng, J.; Gao, N.; Yuan, H.X.; Ye, D.; Guan, K.L.; et al. Structural insights into TSC complex assembly and GAP activity on Rheb. Nat. Commun. 2021, 12, 339. [Google Scholar] [CrossRef]
- Hodges, A.K.; Li, S.; Maynard, J.; Parry, L.; Braverman, R.; Cheadle, J.P.; DeClue, J.E.; Sampson, J.R. Pathological mutations in TSC1 and TSC2 disrupt the interaction between hamartin and tuberin. Hum. Mol. Genet. 2001, 10, 2899–2905. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [Green Version]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, G.; Li, S.; Brown, S.J.; Braverman, R.; Vass, W.C.; Cheadle, J.P.; Halley, D.J.; Sampson, J.R.; Wienecke, R.; DeClue, J.E. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000, 19, 6306–6316. [Google Scholar] [CrossRef] [Green Version]
- Chong-Kopera, H.; Inoki, K.; Li, Y.; Zhu, T.; Garcia-Gonzalo, F.R.; Rosa, J.L.; Guan, K.L. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 2006, 281, 8313–8316. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Kobayashi, T.; Momose, S.; Yasui, H.; Hino, O. Distribution of Tsc1 protein detected by immunohistochemistry in various normal rat tissues and the renal carcinomas of Eker rat: Detection of limited colocalization with Tsc1 and Tsc2 gene products in vivo. Lab. Investig. 2000, 80, 1347–1359. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.J.; Zhang, H.; Bandura, J.L.; Heiberger, K.M.; Glogauer, M.; el-Hashemite, N.; Onda, H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 2002, 11, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; Beaure d’Augeres, G.; de Vries, P.J.; et al. Renal Manifestations of Tuberous Sclerosis Complex: Key Findings from the Final Analysis of the TOSCA Study Focussing Mainly on Renal Angiomyolipomas. Front. Neurol. 2020, 11, 972. [Google Scholar] [CrossRef]
- Qin, Z.; Zheng, H.; Zhou, L.; Ou, Y.; Huang, B.; Yan, B.; Qin, Z.; Yang, C.; Su, Y.; Bai, X.; et al. Tsc1 deficiency impairs mammary development in mice by suppression of AKT, nuclear ERalpha, and cell-cycle-driving proteins. Sci. Rep. 2016, 6, 19587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Diaz, V.; Walsh, M.E.; Zhang, Y. Moderate lifelong overexpression of tuberous sclerosis complex 1 (TSC1) improves health and survival in mice. Sci. Rep. 2017, 7, 834. [Google Scholar] [CrossRef] [Green Version]
- Miloloza, A.; Kubista, M.; Rosner, M.; Hengstschlager, M. Evidence for separable functions of tuberous sclerosis gene products in mammalian cell cycle regulation. J. Neuropathol. Exp. Neurol. 2002, 61, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Thien, A.; Prentzell, M.T.; Holzwarth, B.; Klasener, K.; Kuper, I.; Boehlke, C.; Sonntag, A.G.; Ruf, S.; Maerz, L.; Nitschke, R.; et al. TSC1 activates TGF-beta-Smad2/3 signaling in growth arrest and epithelial-to-mesenchymal transition. Dev. Cell 2015, 32, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Hengstschlager, M.; Rosner, M.; Fountoulakis, M.; Lubec, G. The cellular response to ectopic overexpression of the tuberous sclerosis genes, TSC1 and TSC2: A proteomic approach. Int. J. Oncol. 2005, 27, 831–838. [Google Scholar]
- Rosner, M.; Freilinger, A.; Lubec, G.; Hengstschlager, M. The tuberous sclerosis genes, TSC1 and TSC2, trigger different gene expression responses. Int. J. Oncol. 2005, 27, 1411–1424. [Google Scholar] [CrossRef]
- Dalal, J.S.; Winden, K.D.; Salussolia, C.L.; Sundberg, M.; Singh, A.; Pham, T.T.; Zhou, P.; Pu, W.T.; Miller, M.T.; Sahin, M. Loss of Tsc1 in cerebellar Purkinje cells induces transcriptional and translation changes in FMRP target transcripts. eLife 2021, 10, e67399. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, Q.; Zhang, Z.; Sun, L.; Dong, X.; Li, T.; Tan, L.; Xie, X.; Sun, L.; Zhao, Y. The Development and Survival of Thymic Epithelial Cells Require TSC1-Dependent Negative Regulation of mTORC1 Activity. J. Immunol. 2021, 207, 2039–2050. [Google Scholar] [CrossRef]
- Liu, Y.D.; Ma, M.Y.; Hu, X.B.; Yan, H.; Zhang, Y.K.; Yang, H.X.; Feng, J.H.; Wang, L.; Zhang, H.; Zhang, B.; et al. Brain Proteomic Profiling in Intractable Epilepsy Caused by TSC1 Truncating Mutations: A Small Sample Study. Front. Neurol. 2020, 11, 475. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Zadjali, F.; Yao, Y.; Siroky, B.; Astrinidis, A.; Gross, K.W.; Bissler, J.J. Tsc Gene Locus Disruption and Differences in Renal Epithelial Extracellular Vesicles. Front. Physiol. 2021, 12, 630933. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Bonnet, C.; Guy, C.; Idziaszczyk, S.; Colley, J.; Humphreys, V.; Maynard, J.; Sampson, J.R.; Cheadle, J.P. Tsc1 haploinsufficiency without mammalian target of rapamycin activation is sufficient for renal cyst formation in Tsc1+/− mice. Cancer Res. 2006, 66, 7934–7938. [Google Scholar] [CrossRef] [Green Version]
- Alves, M.M.; Fuhler, G.M.; Queiroz, K.C.; Scholma, J.; Goorden, S.; Anink, J.; Spek, C.A.; Hoogeveen-Westerveld, M.; Bruno, M.J.; Nellist, M.; et al. PAK2 is an effector of TSC1/2 signaling independent of mTOR and a potential therapeutic target for Tuberous Sclerosis Complex. Sci. Rep. 2015, 5, 14534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Goncharova, E.A.; James, M.L.; Kudryashova, T.V.; Goncharov, D.A.; Krymskaya, V.P. Tumor suppressors TSC1 and TSC2 differentially modulate actin cytoskeleton and motility of mouse embryonic fibroblasts. PLoS ONE 2014, 9, e111476. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Zou, W.; Han, Z.; Zhou, L.; Qiu, Z.; Chen, J.; Zhang, S.; Lai, P.; Li, K.; Zhang, Y.; et al. Tsc1 regulates tight junction independent of mTORC1. Proc. Natl. Acad. Sci. USA 2021, 118, e2020891118. [Google Scholar] [CrossRef]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef] [Green Version]
- Curatolo, P.; Moavero, R.; Roberto, D.; Graziola, F. Genotype/Phenotype Correlations in Tuberous Sclerosis Complex. Semin. Pediatr. Neurol. 2015, 22, 259–273. [Google Scholar] [CrossRef]
- Sancak, O.; Nellist, M.; Goedbloed, M.; Elfferich, P.; Wouters, C.; Maat-Kievit, A.; Zonnenberg, B.; Verhoef, S.; Halley, D.; van den Ouweland, A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: Genotype—Phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 2005, 13, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.C.; Daniells, C.E.; Snell, R.G.; Tachataki, M.; Idziaszczyk, S.A.; Krawczak, M.; Sampson, J.R.; Cheadle, J.P. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum. Mol. Genet. 1997, 6, 2155–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niida, Y.; Wakisaka, A.; Tsuji, T.; Yamada, H.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J. Hum. Genet. 2013, 58, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.T.; McCartney, D.L.; Lewis, J.C.; Sampson, J.R.; Howe, C.J.; de Vries, P.J. Intellectual ability in tuberous sclerosis complex correlates with predicted effects of mutations on TSC1 and TSC2 proteins. J. Med. Genet. 2015, 52, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, F.E.; Braams, O.; Vincken, K.L.; Algra, A.; Anbeek, P.; Jennekens-Schinkel, A.; Halley, D.; Zonnenberg, B.A.; van den Ouweland, A.; van Huffelen, A.C.; et al. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology 2008, 70, 908–915. [Google Scholar] [CrossRef]
- Kothare, S.V.; Singh, K.; Chalifoux, J.R.; Staley, B.A.; Weiner, H.L.; Menzer, K.; Devinsky, O. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia 2014, 55, 1025–1029. [Google Scholar] [CrossRef]
- Zeng, L.H.; Rensing, N.R.; Zhang, B.; Gutmann, D.H.; Gambello, M.J.; Wong, M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2011, 20, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Langkau, N.; Martin, N.; Brandt, R.; Zugge, K.; Quast, S.; Wiegele, G.; Jauch, A.; Rehm, M.; Kuhl, A.; Mack-Vetter, M.; et al. TSC1 and TSC2 mutations in tuberous sclerosis, the associated phenotypes and a model to explain observed TSC1/TSC2 frequency ratios. Eur. J. Pediatr. 2002, 161, 393–402. [Google Scholar] [CrossRef]
- Dean, M.E.; Johnson, J.L. Human Hsp90 cochaperones: Perspectives on tissue-specific expression and identification of cochaperones with similar in vivo functions. Cell Stress Chaperones 2021, 26, 3–13. [Google Scholar] [CrossRef]
- Wang, R.Y.; Noddings, C.M.; Kirschke, E.; Myasnikov, A.G.; Johnson, J.L.; Agard, D.A. Structure of Hsp90-Hsp70-Hop-GR reveals the Hsp90 client-loading mechanism. Nature 2022, 601, 460–464. [Google Scholar] [CrossRef]
- Li, J.; Richter, K.; Reinstein, J.; Buchner, J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 326–331. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.; Dahiya, V.; Delhommel, F.; Freiburger, L.; Stehle, R.; Asami, S.; Rutz, D.; Blair, L.; Buchner, J.; Sattler, M. Client binding shifts the populations of dynamic Hsp90 conformations through an allosteric network. Sci. Adv. 2021, 7, eabl7295. [Google Scholar] [CrossRef]
- Biebl, M.M.; Riedl, M.; Buchner, J. Hsp90 Co-chaperones Form Plastic Genetic Networks Adapted to Client Maturation. Cell Rep. 2020, 32, 108063. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Mollapour, M.; Smith, J.R.; Truman, A.; Hu, B.; Good, V.M.; Panaretou, B.; Neckers, L.; Clarke, P.A.; Workman, P.; et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 2008, 31, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Sager, R.A.; Dushukyan, N.; Woodford, M.; Mollapour, M. Structure and function of the co-chaperone protein phosphatase 5 in cancer. Cell Stress Chaperones 2020, 25, 383–394. [Google Scholar] [CrossRef]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Koulov, A.V.; LaPointe, P.; Lu, B.; Razvi, A.; Coppinger, J.; Dong, M.Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R., 3rd; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar] [CrossRef]
- Dunn, D.M.; Woodford, M.R.; Truman, A.W.; Jensen, S.M.; Schulman, J.; Caza, T.; Remillard, T.C.; Loiselle, D.; Wolfgeher, D.; Blagg, B.S.; et al. c-Abl Mediated Tyrosine Phosphorylation of Aha1 Activates Its Co-chaperone Function in Cancer Cells. Cell Rep. 2015, 12, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef] [Green Version]
- Rohl, A.; Tippel, F.; Bender, E.; Schmid, A.B.; Richter, K.; Madl, T.; Buchner, J. Hop/Sti1 phosphorylation inhibits its co-chaperone function. EMBO Rep. 2015, 16, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohl, A.; Wengler, D.; Madl, T.; Lagleder, S.; Tippel, F.; Herrmann, M.; Hendrix, J.; Richter, K.; Hack, G.; Schmid, A.B.; et al. Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 2015, 6, 6655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, O.R.; Freiburger, L.; Rutz, D.A.; Krause, M.; Zierer, B.K.; Alvira, S.; Cuellar, J.; Valpuesta, J.M.; Madl, T.; Sattler, M.; et al. Modulation of the Hsp90 chaperone cycle by a stringent client protein. Mol. Cell 2014, 53, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Sager, R.A.; Woodford, M.R.; Backe, S.J.; Makedon, A.M.; Baker-Williams, A.J.; DiGregorio, B.T.; Loiselle, D.R.; Haystead, T.A.; Zachara, N.E.; Prodromou, C.; et al. Post-translational Regulation of FNIP1 Creates a Rheostat for the Molecular Chaperone Hsp90. Cell Rep. 2019, 26, 1344–1356.e1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, N.; Shaik, A.; Thiruvenkatam, V. Recombinant Tumor Suppressor TSC1 Differentially Interacts with Escherichia coli DnaK and Human HSP70. ACS Omega 2020, 5, 19131–19139. [Google Scholar] [CrossRef] [PubMed]
- Hessling, M.; Richter, K.; Buchner, J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2009, 16, 287–293. [Google Scholar] [CrossRef]
- Mickler, M.; Hessling, M.; Ratzke, C.; Buchner, J.; Hugel, T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 2009, 16, 281–286. [Google Scholar] [CrossRef]
- Prodromou, C.; Pearl, L.H. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Noddings, C.M.; Wang, R.Y.; Johnson, J.L.; Agard, D.A. Structure of Hsp90-p23-GR reveals the Hsp90 client-remodelling mechanism. Nature 2022, 601, 465–469. [Google Scholar] [CrossRef]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blacklock, K.; Verkhivker, G.M. Differential modulation of functional dynamics and allosteric interactions in the Hsp90-cochaperone complexes with p23 and Aha1: A computational study. PLoS ONE 2013, 8, e71936. [Google Scholar] [CrossRef] [PubMed]
- Cano, L.Q.; Lavery, D.N.; Sin, S.; Spanjaard, E.; Brooke, G.N.; Tilman, J.D.; Abroaf, A.; Gaughan, L.; Robson, C.N.; Heer, R.; et al. The co-chaperone p23 promotes prostate cancer motility and metastasis. Mol. Oncol. 2015, 9, 295–308. [Google Scholar] [CrossRef]
- Martinez-Yamout, M.A.; Venkitakrishnan, R.P.; Preece, N.E.; Kroon, G.; Wright, P.E.; Dyson, H.J. Localization of sites of interaction between p23 and Hsp90 in solution. J. Biol. Chem. 2006, 281, 14457–14464. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, S.H.; Sobott, F.; Yao, Z.P.; Zhang, W.; Nielsen, P.R.; Grossmann, J.G.; Laue, E.D.; Robinson, C.V.; Jackson, S.E. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J. Mol. Biol. 2006, 356, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Walter, S.; Buchner, J. The Co-chaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J. Mol. Biol. 2004, 342, 1403–1413. [Google Scholar] [CrossRef]
- Woo, S.H.; An, S.; Lee, H.C.; Jin, H.O.; Seo, S.K.; Yoo, D.H.; Lee, K.H.; Rhee, C.H.; Choi, E.J.; Hong, S.I.; et al. A truncated form of p23 down-regulates telomerase activity via disruption of Hsp90 function. J. Biol. Chem. 2009, 284, 30871–30880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Woodford, M.R.; Hughes, M.; Sager, R.A.; Backe, S.J.; Baker-Williams, A.J.; Bratslavsky, M.S.; Jacob, J.M.; Shapiro, O.; Wong, M.; Bratslavsky, G.; et al. Mutation of the co-chaperone Tsc1 in bladder cancer diminishes Hsp90 acetylation and reduces drug sensitivity and selectivity. Oncotarget 2019, 10, 5824–5834. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Truman, A.W.; Dunn, D.M.; Jensen, S.M.; Cotran, R.; Bullard, R.; Abouelleil, M.; Beebe, K.; Wolfgeher, D.; Wierzbicki, S.; et al. Mps1 Mediated Phosphorylation of Hsp90 Confers Renal Cell Carcinoma Sensitivity and Selectivity to Hsp90 Inhibitors. Cell Rep. 2016, 14, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.J.; et al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell 2014, 53, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisht, K.S.; Bradbury, C.M.; Mattson, D.; Kaushal, A.; Sowers, A.; Markovina, S.; Ortiz, K.L.; Sieck, L.K.; Isaacs, J.S.; Brechbiel, M.W.; et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 2003, 63, 8984–8995. [Google Scholar]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yi, Y.; Guo, Q.; Sun, Y.; Ma, S.; Xiao, S.; Geng, J.; Zheng, Z.; Song, S. Hsp90 interacts with AMPK and mediates acetyl-CoA carboxylase phosphorylation. Cell Signal. 2012, 24, 859–865. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, K.; Fukushima, H.; Shimizu, K.; Yamada, A.; Hidaka, M.; Hasumi, H.; Ikebe, T.; Fukumoto, S.; Okabe, K.; Inuzuka, H. Nutrient-induced FNIP degradation by SCFbeta-TRCP regulates FLCN complex localization and promotes renal cancer progression. Oncotarget 2017, 8, 9947–9960. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Horejsi, Z.; Takai, H.; Adelman, C.A.; Collis, S.J.; Flynn, H.; Maslen, S.; Skehel, J.M.; de Lange, T.; Boulton, S.J. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 2010, 39, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Miao, S.; Zhang, B.; He, S.; Shou, C.; Manivel, P.; Krishna, R.; Chen, Y.; Shi, Y.E. Synuclein gamma protects Akt and mTOR and renders tumor resistance to Hsp90 disruption. Oncogene 2015, 34, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Ohji, G.; Hidayat, S.; Nakashima, A.; Tokunaga, C.; Oshiro, N.; Yoshino, K.I.; Yokono, K.; Kikkawa, U.; Yonezawa, K. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J. Biochem. 2006, 139, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Xie, Y.; de Lange, T.; Pavletich, N.P. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 2010, 24, 2019–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquaviva, J.; He, S.; Zhang, C.; Jimenez, J.P.; Nagai, M.; Sang, J.; Sequeira, M.; Smith, D.L.; Ogawa, L.S.; Inoue, T.; et al. FGFR3 translocations in bladder cancer: Differential sensitivity to HSP90 inhibition based on drug metabolism. Mol. Cancer Res. 2014, 12, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Giulino-Roth, L.; van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of Hsp90 Suppresses PI3K/AKT/mTOR Signaling and Has Antitumor Activity in Burkitt Lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Solarova, Z.; Mojzis, J.; Solar, P. Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int. J. Oncol. 2015, 46, 907–926. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, A.; Lenoel, I.; Winden, K.D.; Ruhmkorf, A.; Modi, M.E.; Barrett, L.; Ercan-Herbst, E.; Venugopal, P.; Behne, R.; Lopes, C.A.M.; et al. Phenotypic Screen with TSC-Deficient Neurons Reveals Heat-Shock Machinery as a Druggable Pathway for mTORC1 and Reduced Cilia. Cell Rep. 2020, 31, 107780. [Google Scholar] [CrossRef]
- Mrozek, E.M.; Bajaj, V.; Guo, Y.; Malinowska, I.A.; Zhang, J.; Kwiatkowski, D.J. Evaluation of Hsp90 and mTOR inhibitors as potential drugs for the treatment of TSC1/TSC2 deficient cancer. PLoS ONE 2021, 16, e0248380. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Sutphin, P.D.; Pulcini, E.J.; Martinez, J.D.; Cook, P.H. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol. Cell Biol. 1998, 18, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; Schaupp, A.; Walerych, D.; Wegele, H.; Buchner, J. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J. Biol. Chem. 2004, 279, 48846–48854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walerych, D.; Kudla, G.; Gutkowska, M.; Wawrzynow, B.; Muller, L.; King, F.W.; Helwak, A.; Boros, J.; Zylicz, A.; Zylicz, M. Hsp90 chaperones wild-type p53 tumor suppressor protein. J. Biol. Chem. 2004, 279, 48836–48845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennisi, R.; Antoccia, A.; Leone, S.; Ascenzi, P.; di Masi, A. Hsp90alpha regulates ATM and NBN functions in sensing and repair of DNA double-strand breaks. FEBS J. 2017, 284, 2378–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudeau, J.; Deak, M.; Lawlor, M.A.; Morrice, N.A.; Alessi, D.R. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem. J. 2003, 370, 849–857. [Google Scholar] [CrossRef]
- Walter, R.; Pan, K.T.; Doebele, C.; Comoglio, F.; Tomska, K.; Bohnenberger, H.; Young, R.M.; Jacobs, L.; Keller, U.; Bonig, H.; et al. HSP90 promotes Burkitt lymphoma cell survival by maintaining tonic B-cell receptor signaling. Blood 2017, 129, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Narayan, V.; Eckert, M.; Zylicz, A.; Zylicz, M.; Ball, K.L. Cooperative regulation of the interferon regulatory factor-1 tumor suppressor protein by core components of the molecular chaperone machinery. J. Biol. Chem. 2009, 284, 25889–25899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, H.; Bansal, S.; Rao, M.; Foley, K.P.; Sang, J.; Proia, D.A.; Blackman, R.K.; Ying, W.; Barsoum, J.; Baer, M.R.; et al. Heat shock protein 90 regulates the expression of Wilms tumor 1 protein in myeloid leukemias. Blood 2010, 116, 4591–4599. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef]
- Manjarrez, J.R.; Sun, L.; Prince, T.; Matts, R.L. Hsp90-dependent assembly of the DBC2/RhoBTB2-Cullin3 E3-ligase complex. PLoS ONE 2014, 9, e90054. [Google Scholar] [CrossRef] [Green Version]
- McClellan, A.J.; Scott, M.D.; Frydman, J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 2005, 121, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntoon, C.J.; Nye, M.D.; Geng, L.; Peterson, K.L.; Flatten, K.S.; Haluska, P.; Kaufmann, S.H.; Karnitz, L.M. Heat shock protein 90 inhibition depletes LATS1 and LATS2, two regulators of the mammalian hippo tumor suppressor pathway. Cancer Res. 2010, 70, 8642–8650. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Shanab, O.; Nakahata, S.; Shimosaki, S.; Manachai, N.; Ono, M.; Iha, H.; Shimoda, K.; Morishita, K. Novel PRMT5-mediated arginine methylations of HSP90A are essential for maintenance of HSP90A function in NDRG2(low) ATL and various cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118615. [Google Scholar] [CrossRef] [PubMed]
- King, F.W.; Wawrzynow, A.; Hohfeld, J.; Zylicz, M. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 2001, 20, 6297–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Gallardo, L.; Martin-Benito, J.; Marcilla, M.; Espadas, G.; Sabido, E.; Valpuesta, J.M. The cochaperone CHIP marks Hsp70- and Hsp90-bound substrates for degradation through a very flexible mechanism. Sci. Rep. 2019, 9, 5102. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Meng, Z.; Moroishi, T.; Lin, K.C.; Shen, G.; Mo, F.; Shao, B.; Wei, X.; Zhang, P.; Wei, Y.; et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat. Cell Biol. 2020, 22, 1447–1459. [Google Scholar] [CrossRef]
- Shen, J.P.; Zhao, D.; Sasik, R.; Luebeck, J.; Birmingham, A.; Bojorquez-Gomez, A.; Licon, K.; Klepper, K.; Pekin, D.; Beckett, A.N.; et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 2017, 14, 573–576. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.N.; Fan, C.C.; Lo, Y.K.; Kuo, C.L.; Wang, H.C.; Lien, I.H.; Lin, S.Y.; Chen, C.H.; Jiang, S.S.; Chang, I.S.; et al. Cdc7-Dbf4-mediated phosphorylation of HSP90-S164 stabilizes HSP90-HCLK2-MRN complex to enhance ATR/ATM signaling that overcomes replication stress in cancer. Sci. Rep. 2017, 7, 17024. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [Green Version]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef] [PubMed]
- Nony, P.; Gaude, H.; Rossel, M.; Fournier, L.; Rouault, J.P.; Billaud, M. Stability of the Peutz-Jeghers syndrome kinase LKB1 requires its binding to the molecular chaperones Hsp90/Cdc37. Oncogene 2003, 22, 9165–9175. [Google Scholar] [CrossRef] [Green Version]
- Gaude, H.; Aznar, N.; Delay, A.; Bres, A.; Buchet-Poyau, K.; Caillat, C.; Vigouroux, A.; Rogon, C.; Woods, A.; Vanacker, J.M.; et al. Molecular chaperone complexes with antagonizing activities regulate stability and activity of the tumor suppressor LKB1. Oncogene 2012, 31, 1582–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, A.; Lu, P.; Lee, J.; Zhen, C.; Chiosis, G.; Wang, Y.L. HSP90 stabilizes B-cell receptor kinases in a multi-client interactome: PU-H71 induces CLL apoptosis in a cytoprotective microenvironment. Oncogene 2017, 36, 3441–3449. [Google Scholar] [CrossRef]
- Shen, L.J.; Sun, H.W.; Chai, Y.Y.; Jiang, Q.Y.; Zhang, J.; Li, W.M.; Xin, S.J. The Disassociation of the A20/HSP90 Complex via Downregulation of HSP90 Restores the Effect of A20 Enhancing the Sensitivity of Hepatocellular Carcinoma Cells to Molecular Targeted Agents. Front. Oncol. 2021, 11, 804412. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated Conformational Processing of the Tumor Suppressor Protein p53 by the Hsp70 and Hsp90 Chaperone Machineries. Mol. Cell 2019, 74, 816–830.e817. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, R.; Kiang, J.G. Geldanamycin analog 17-DMAG limits apoptosis in human peripheral blood cells by inhibition of p53 activation and its interaction with heat-shock protein 90 kDa after exposure to ionizing radiation. Radiat. Res. 2011, 176, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustiniani, J.; Daire, V.; Cantaloube, I.; Durand, G.; Pous, C.; Perdiz, D.; Baillet, A. Tubulin acetylation favors Hsp90 recruitment to microtubules and stimulates the signaling function of the Hsp90 clients Akt/PKB and p53. Cell Signal. 2009, 21, 529–539. [Google Scholar] [CrossRef]
- Hagn, F.; Lagleder, S.; Retzlaff, M.; Rohrberg, J.; Demmer, O.; Richter, K.; Buchner, J.; Kessler, H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat. Struct. Mol. Biol. 2011, 18, 1086–1093. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Anan, T.; Yoshida, T.; Mizukami, T.; Taya, Y.; Fujiwara, T.; Kato, H.; Saya, H.; Nakao, M. The stabilization mechanism of mutant-type p53 by impaired ubiquitination: The loss of wild-type p53 function and the hsp90 association. Oncogene 1999, 18, 6037–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Borin, B.N.; Martinez-Yamout, M.A.; Dyson, H.J. The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat. Struct. Mol. Biol. 2011, 18, 537–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Kostic, M.; Dyson, H.J. Dynamic Interaction of Hsp90 with Its Client Protein p53. J. Mol. Biol. 2011, 411, 158–173. [Google Scholar] [CrossRef] [Green Version]
- Rudiger, S.; Freund, S.M.; Veprintsev, D.B.; Fersht, A.R. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl. Acad. Sci. USA 2002, 99, 11085–11090. [Google Scholar] [CrossRef] [Green Version]
- Walerych, D.; Gutkowska, M.; Klejman, M.P.; Wawrzynow, B.; Tracz, Z.; Wiech, M.; Zylicz, M.; Zylicz, A. ATP binding to Hsp90 is sufficient for effective chaperoning of p53 protein. J. Biol. Chem. 2010, 285, 32020–32028. [Google Scholar] [CrossRef] [Green Version]
- Denney, A.S.; Weems, A.D.; McMurray, M.A. Selective functional inhibition of a tumor-derived p53 mutant by cytosolic chaperones identified using split-YFP in budding yeast. G3 2021, 11, jkab230. [Google Scholar] [CrossRef]
- Woodford, M.R.; Backe, S.J.; Wengert, L.A.; Dunn, D.M.; Bourboulia, D.; Mollapour, M. Hsp90 chaperone code and the tumor suppressor VHL cooperatively regulate the mitotic checkpoint. Cell Stress Chaperones 2021, 26, 965–971. [Google Scholar] [CrossRef]
- Centini, R.; Tsang, M.; Iwata, T.; Park, H.; Delrow, J.; Margineantu, D.; Iritani, B.M.; Gu, H.; Liggitt, H.D.; Kang, J.; et al. Loss of Fnip1 alters kidney developmental transcriptional program and synergizes with TSC1 loss to promote mTORC1 activation and renal cyst formation. PLoS ONE 2018, 13, e0197973. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Suppressor Gene | Relationship to Hsp90 | References |
|---|---|---|
| ATM Kinase | Client | [153,169] |
| BMPR1A | Client | [161] |
| BRCA1/2 | Client | [158,170,171] |
| DBC2 | Client | [159] |
| FBXW7 | Interactor | [161] |
| FLCN | Client | [20,23] |
| IRF1 | Client | [156] |
| LATS1/2 | Client | [162,167,172] |
| LKB1 | Client | [154,173,174] |
| NDRG2 | Interactor | [163] |
| SYK | Client | [155,175] |
| TNFAIP3 | Interactor | [161,176] |
| TP53 | Client | [8,103,149,150,151,152,164,165,166,177,178,179,180,181,182,183,184,185,186,187] |
| TSC1 | Co-chaperone | [21,130] |
| TSC2 | Client | [21] |
| VHL | Client | [160,188] |
| WT1 | Client | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Backe, S.J.; Sager, R.A.; Meluni, K.A.; Woodford, M.R.; Bourboulia, D.; Mollapour, M. Emerging Link between Tsc1 and FNIP Co-Chaperones of Hsp90 and Cancer. Biomolecules 2022, 12, 928. https://doi.org/10.3390/biom12070928
Backe SJ, Sager RA, Meluni KA, Woodford MR, Bourboulia D, Mollapour M. Emerging Link between Tsc1 and FNIP Co-Chaperones of Hsp90 and Cancer. Biomolecules. 2022; 12(7):928. https://doi.org/10.3390/biom12070928
Chicago/Turabian StyleBacke, Sarah J., Rebecca A. Sager, Katherine A. Meluni, Mark R. Woodford, Dimitra Bourboulia, and Mehdi Mollapour. 2022. "Emerging Link between Tsc1 and FNIP Co-Chaperones of Hsp90 and Cancer" Biomolecules 12, no. 7: 928. https://doi.org/10.3390/biom12070928
APA StyleBacke, S. J., Sager, R. A., Meluni, K. A., Woodford, M. R., Bourboulia, D., & Mollapour, M. (2022). Emerging Link between Tsc1 and FNIP Co-Chaperones of Hsp90 and Cancer. Biomolecules, 12(7), 928. https://doi.org/10.3390/biom12070928