_Kwok.png)
Molecular Determinants Underlying the Anti-Cancer Efficacy of CD38 Monoclonal Antibodies in Hematological Malignancies

Abstract
:1. Introduction
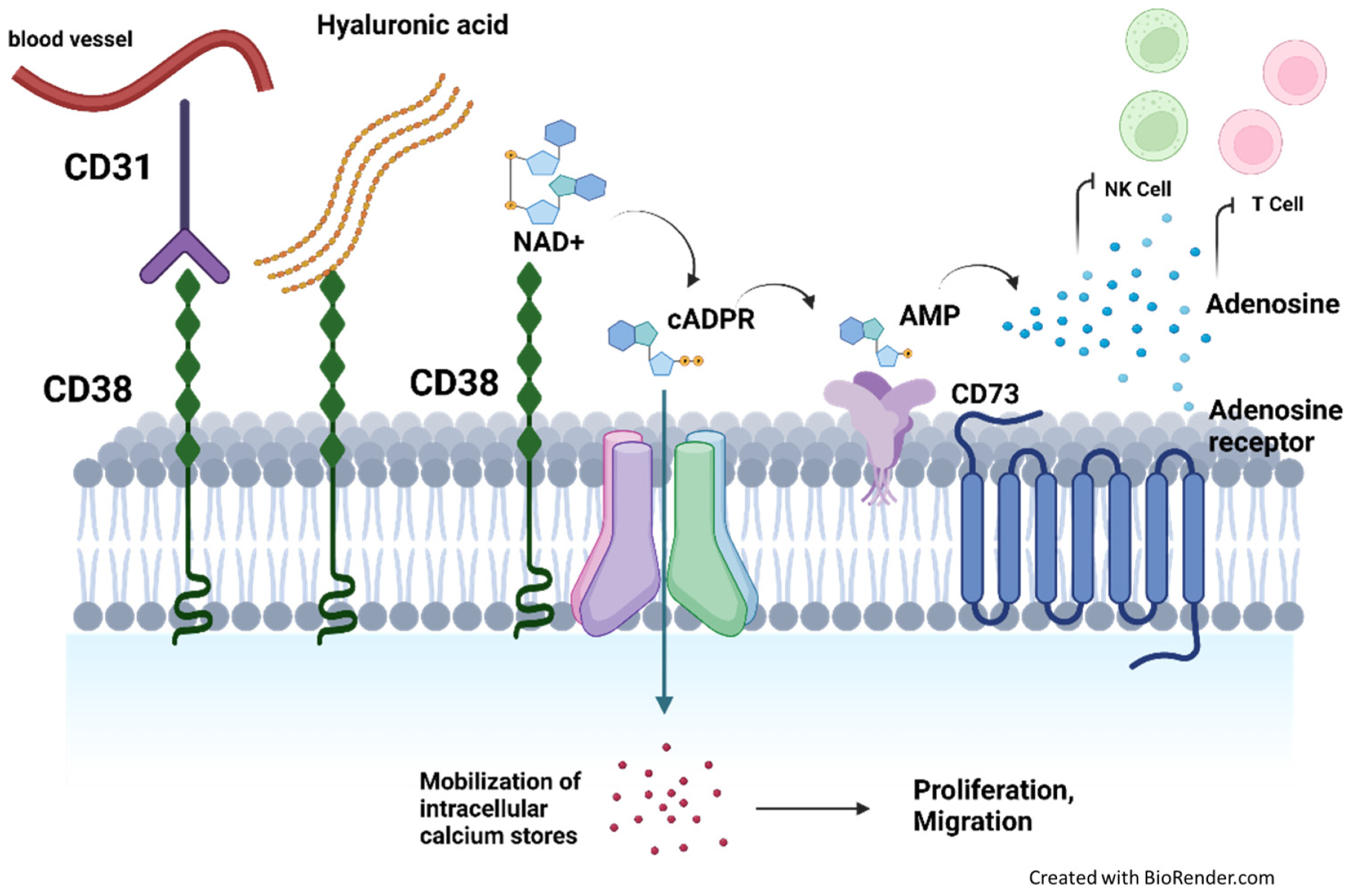
2. CD38 Protein Expression and Function in Healthy Cells
3. CD38-Mediated Tumor-Promoting Mechanisms and Expression in Hematological Cancers
3.1. CD38 in Multiple Myeloma
3.1.1. CD38 Increases Capacity for Oxidative Phosphorylation
3.1.2. CD38 Mediates Immunosuppression by Producing Elevated Levels of Adenosine
3.1.3. CD38 Expression on Immune Suppressor Cells Promote a Pro-Tumoral MM Niche
3.2. CD38 in Chronic Lymphocytic Leukemia
3.2.1. CD38 Promotes Migration towards Proliferative Niches through Adhesion and Cytokine Production
3.2.2. CD38 Directly Stimulates Growth and Survival Signals
3.2.3. CD38+ CLL Cells Exhibit Intrinsic Molecular Characteristics in Favor of Promoting Oncogenesis
3.2.4. CD38 Expression on Immune Suppressor Cells Promote a Pro-Tumoral CLL Niche
3.3. CD38 in Aggressive Non-Hodgkin Lymphomas
3.4. CD38 in T and NK Lymphomas

{kind=link}
{kind=link}
{kind=link}
| CD38 Function | How This Is Hijacked to Promote Cancer | Type of Blood Malignancy |
|---|---|---|
| Ecto-enzymatic NADase activity | Elevated levels of adenosine suppress activity of effector immune cells and stimulate activity of regulatory T cells and myeloid-derived suppressor cells | MM, DLBCL, T-ALL |
| Increased enzymatic activity increases production of cADPR and NAADP calcium messengers, which promote survival, trafficking, and homing | MM, CLL, AML | |
| Adhesion | Formation of nanotubes to mediate mitochondrial transfer from BMSC to promote oxidative phosphorylation | MM |
| Cell surface receptor/antigen | Increased expression on immune suppressor cells, which intensifies cell suppressive phenotype and promotes formation of immune-suppressive tumor niches | MM, CLL |
| Chemokine-mediated migration towards proliferative niches | CLL, AML | |
| Colocalization with other receptors to directly transduce survival signaling | CLL | |
| Biomarker for poor prognosis | CLL, MCL, DLBCL, PTCL, NKTL |
3.5. CD38 in Acute Myeloid Leukemia (AML) and T cell Acute Lymphoblastic Leukemia (T-ALL)
3.6. Clinical Studies of CD38-Targeting Antibodies in Hematological Malignancies (Table 2)
| Tumor Type | Study Title | Phase | Drug Regimen | Median PFS | Ref |
|---|---|---|---|---|---|
| MM | NCT02076009, POLLUX | 3 | Dara-Len-Dex vs. Len-Dex | 44.5 vs. 17.5 months | [85,86] |
| NCT03180736, APOLLO | 3 | Dara-Pom-Dex vs. Pom-Dex | 12.4 vs. 6.9 months | [87] | |
| NCT02136134, CASTOR | 3 | Dara-Bort-Dex vs. Bort-Dex | 60.7 vs. 26.9 months | [5,93] | |
| NCT03158688, CANDOR | 3 | Dara-Carfil-Dex vs. Carfil-Dex | 28.6 vs. 15.2 months | [91] | |
| NCT01749969 | 1b | Isa-Len-Dex | 8.5 months | [88] | |
| NCT02990338, ICARIA-MM | 3 | Isa-Pom-Dex vs. Pom-Dex | 11.5 vs. 6.5 months | [9] | |
| NCT03275285, IKEMA | 3 | Isa-Carfilz-Dex vs. Carfilz-Dex | 35.7 vs. 19.2 months | [10] | |
| NCT01421186 | 1b/2a | MOR202-Len-Dex | not reached after 24 months | [11] | |
| NCT01421186 | 1b/2a | MOR202-Pom-Dex vs. Mor Dex | 17.5 vs. 8.4 months | [11] | |
| NKTL | NCT02927925 | 2 | Dara single agent | 55 days | [21] |
| MCL, DLBCL, FL | NCT02413489, CARINA | 2 | Dara single agent | Terminated as futility thresholds were not reached (FL ORR 50%), (DLBCL ORR 30%) | [22] |
| T ALL, TLBL | NCT02999633 | 2 | Isa single agent | Terminated; unsatisfactory benefit/risk ratio, 11/14 developed progressive disease as best response. | [92] |
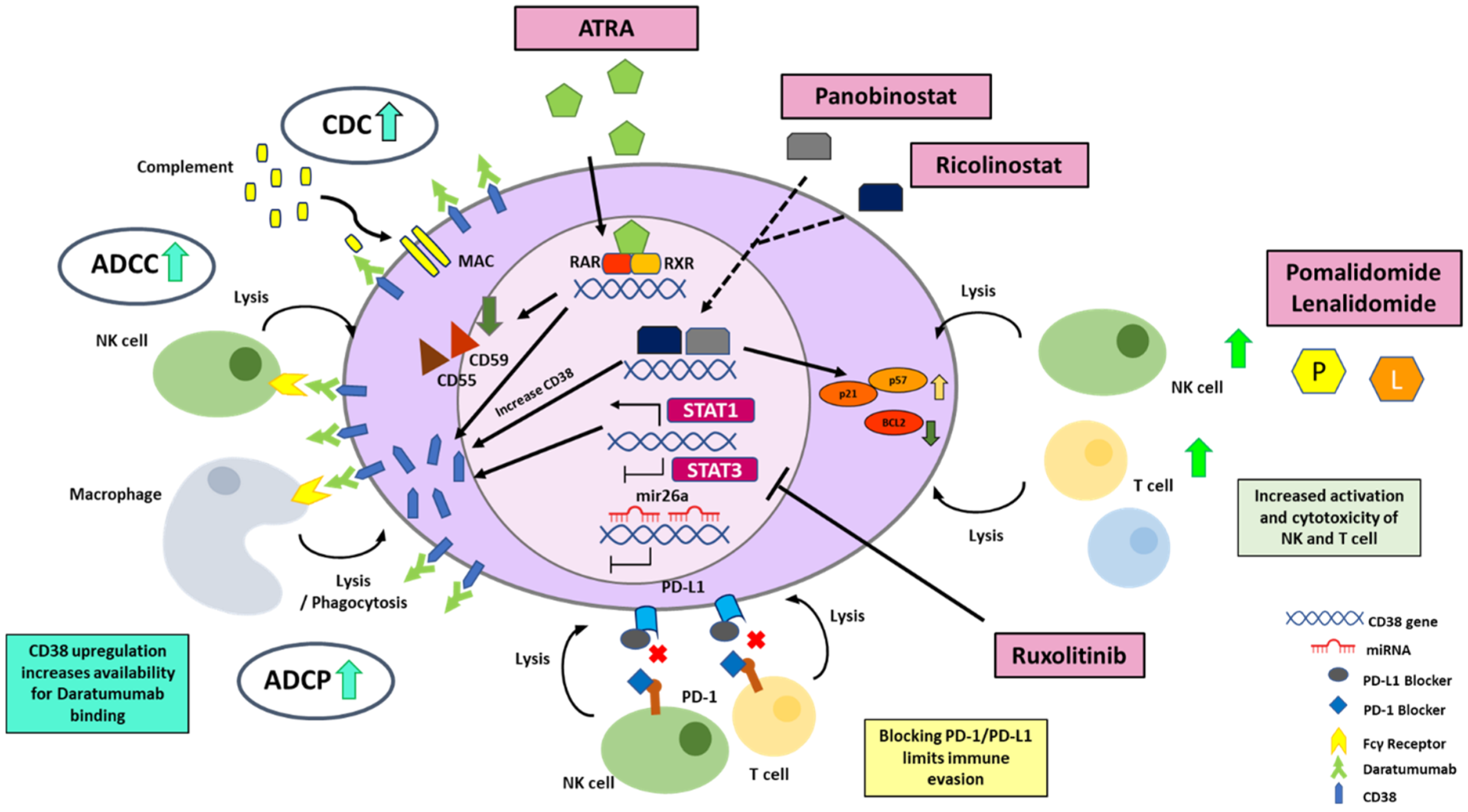
4. Molecular Strategies to Enhance CD38 Expression for More Effective Targeting by Monoclonal Antibodies
4.1. The Human CD38 Gene
4.2. Strategies to Enhance Transcriptional Activation of CD38 Gene
4.2.1. All Trans Retinoic Acid (ATRA)
4.2.2. HDAC Inhibitors
4.2.3. STAT 3 Inhibitors
4.2.4. Immunomodulatory Imide Drugs (IMiDs)
4.3. Strategies Regulating the Degradation of CD38 mRNA
4.4. Strategies to Optimize CD38 Antigen Availability on the Cell Membrane
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.K. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Bene, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Postigo, J.; Iglesias, M.; Cerezo-Wallis, D.; Rosal-Vela, A.; García-Rodríguez, S.; Zubiaur, M.; Sancho, J.; Merino, R.; Merino, J. Mice Deficient in CD38 Develop an Attenuated Form of Collagen Type II-Induced Arthritis. PLoS ONE 2012, 7, e33534. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef]
- van Bueren, J.L.; Jakobs, D.; Kaldenhoven, N.; Roza, M.; Hiddingh, S.; Meesters, J.; Voorhorst, M.; Gresnigt, E.; Wiegman, L.; Buijsse, A.O.; et al. Direct in Vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood 2014, 124, 3474. [Google Scholar] [CrossRef]
- Richardson, P.G.; Perrot, A.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; Huang, J.S.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): Follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022, 23, 416–427. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Spicka, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Rollig, C.; et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: A first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef] [PubMed]
- Fedyk, E.R.; Zhao, L.; Koch, A.; Smithson, G.; Estevam, J.; Chen, G.; Lahu, G.; Roepcke, S.; Lin, J.; McLean, L. Safety, tolerability, pharmacokinetics and pharmacodynamics of the anti-CD38 cytolytic antibody TAK-079 in healthy subjects. Br. J. Clin. Pharmacol. 2020, 86, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Wada, F.; Shimomura, Y.; Yabushita, T.; Yamashita, D.; Ohno, A.; Imoto, H.; Maruoka, H.; Hara, S.; Ishikawa, T. CD38 expression is an important prognostic marker in diffuse large B-cell lymphoma. Hematol. Oncol. 2021, 39, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, N.; Nee, A.H.F.; Chooi, J.Y.; Toh, S.H.M.; Chung, T.H.; Selvarajan, V.; Fan, S.; Ng, S.B.; Poon, M.; Chan, E.; et al. Determinants of response to daratumumab in Epstein-Barr virus-positive natural killer and T-cell lymphoma. J. Immunother. Cancer 2021, 9, e002123. [Google Scholar] [CrossRef] [PubMed]
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef]
- Ghia, P.; Guida, G.; Stella, S.; Gottardi, D.; Geuna, M.; Strola, G.; Scielzo, C.; Caligaris-Cappio, F. The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood 2003, 101, 1262–1269. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Bergui, L.; Bonello, L.; Horenstein, A.L.; Tamagnone, L.; Boumsell, L.; Malavasi, F. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood 2005, 105, 3042–3050. [Google Scholar] [CrossRef]
- Vaisitti, T.; Aydin, S.; Rossi, D.; Cottino, F.; Bergui, L.; D’Arena, G.; Bonello, L.; Horenstein, A.L.; Brennan, P.; Pepper, C.; et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia 2010, 24, 958–969. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhu, J.; Yao, M.; Kim, T.M.; Yoon, D.H.; Cho, S.G.; Eom, H.S.; Lim, S.T.; Yeh, S.P.; Song, Y.; et al. Daratumumab monotherapy for patients with relapsed or refractory natural killer/T-cell lymphoma, nasal type: An open-label, single-arm, multicenter, phase 2 study. J. Hematol. Oncol. 2021, 14, 25. [Google Scholar] [CrossRef]
- Salles, G.; Gopal, A.K.; Minnema, M.C.; Wakamiya, K.; Feng, H.; Schecter, J.M.; Wang, M. Phase 2 Study of Daratumumab in Relapsed/Refractory Mantle-Cell Lymphoma, Diffuse Large B-Cell Lymphoma, and Follicular Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 275–284. [Google Scholar] [CrossRef]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Levey, R.H.; Schlossman, S.F. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. USA 1980, 77, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Lund, F.; Solvason, N.; Grimaldi, J.C.; Parkhouse, R.M.; Howard, M. Murine CD38: An immunoregulatory ectoenzyme. Immunol. Today 1995, 16, 469–473. [Google Scholar] [CrossRef]
- Malavasi, F.; Funaro, A.; Roggero, S.; Horenstein, A.; Calosso, L.; Mehta, K. Human CD38: A glycoprotein in search of a function. Immunol. Today 1994, 15, 95–97. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Otsuka, N.; Sato, M.; Ishii, Y.; Kon, S.; Yamada, M.; Nishina, H.; Katada, T.; Ikeda, K. Neuronal localization of CD38 antigen in the human brain. Brain Res. 1995, 697, 235–240. [Google Scholar] [CrossRef]
- Cockayne, D.A.; Muchamuel, T.; Grimaldi, J.C.; Muller-Steffner, H.; Randall, T.D.; Lund, F.E.; Murray, R.; Schuber, F.; Howard, M.C. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 1998, 92, 1324–1333. [Google Scholar] [CrossRef]
- Nishina, H.; Inageda, K.; Takahashi, K.; Hoshino, S.; Ikeda, K.; Katada, T. Cell surface antigen CD38 identified as ecto-enzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochem. Biophys. Res. Commun. 1994, 203, 1318–1323. [Google Scholar] [CrossRef]
- Newman, P.J. Switched at birth: A new family for PECAM-1. J. Clin. Investig. 1999, 103, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Frasca, L.; Fedele, G.; Deaglio, S.; Capuano, C.; Palazzo, R.; Vaisitti, T.; Malavasi, F.; Ausiello, C.M. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 2006, 107, 2392–2399. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Deterre, P.; Berthelier, V.; Bauvois, B.; Dalloul, A.; Schuber, F.; Lund, F. CD38 in T- and B-cell functions. Chem. Immunol. 2000, 75, 146–168. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, E.; Franco, L.; Guida, L.; Benatti, U.; Bargellesi, A.; Malavasi, F.; Lee, H.C.; De Flora, A. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 1993, 196, 1459–1465. [Google Scholar] [CrossRef]
- Lee, H.C.; Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef]
- Schuber, F.; Lund, F.E. Structure and enzymology of ADP-ribosyl cyclases: Conserved enzymes that produce multiple calcium mobilizing metabolites. Curr. Mol. Med. 2004, 4, 249–261. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef]
- Ramakers, B.P.; Wever, K.E.; Kox, M.; van den Broek, P.H.; Mbuyi, F.; Rongen, G.; Masereeuw, R.; van der Hoeven, J.G.; Smits, P.; Riksen, N.P.; et al. How systemic inflammation modulates adenosine metabolism and adenosine receptor expression in humans in vivo. Crit. Care Med. 2012, 40, 2609–2616. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights From Basic Science to Therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcgamma Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Kyle, R.A.; Greipp, P.R. The plasma cell labeling index: A valuable tool in primary systemic amyloidosis. Blood 1989, 74, 1108–1111. [Google Scholar] [CrossRef]
- Muchtar, E.; Jevremovic, D.; Dispenzieri, A.; Dingli, D.; Buadi, F.K.; Lacy, M.Q.; Gonsalves, W.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. The prognostic value of multiparametric flow cytometry in AL amyloidosis at diagnosis and at the end of first-line treatment. Blood 2017, 129, 82–87. [Google Scholar] [CrossRef]
- Kaufman, G.P.; Schrier, S.L.; Lafayette, R.A.; Arai, S.; Witteles, R.M.; Liedtke, M. Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis. Blood 2017, 130, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Di Nora, C.; Sponga, S.; Ferrara, V.; Patriarca, F.; Fanin, R.; Nalli, C.; Lechiancole, A.; Vendramin, I.; Livi, U. Emerging therapy in light-chain and acquired transthyretin-related amyloidosis: An Italian single-centre experience in heart transplantation. J. Cardiovasc. Med. 2021, 22, 261–267. [Google Scholar] [CrossRef]
- Morice, W.G.; Chen, D.; Kurtin, P.J.; Hanson, C.A.; McPhail, E.D. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenstrom’s macroglobulinemia. Mod. Pathol. 2009, 22, 807–816. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Vidriales, M.B.; Ocio, E.; Mateo, G.; Sanchez-Guijo, F.; Sanchez, M.L.; Escribano, L.; Barez, A.; Moro, M.J.; Hernandez, J.; et al. Immunophenotypic analysis of Waldenstrom’s macroglobulinemia. Semin. Oncol. 2003, 30, 187–195. [Google Scholar] [CrossRef]
- Konoplev, S.; Medeiros, L.J.; Bueso-Ramos, C.E.; Jorgensen, J.L.; Lin, P. Immunophenotypic profile of lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Am. J. Clin. Pathol. 2005, 124, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Waldenström Macroglobulinemia Patients with Daratumumab. 2020. Available online: Clinicaltrials.gov (accessed on 30 July 2022).
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.B.; Lee, J.S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef]
- Patten, P.E.; Buggins, A.G.; Richards, J.; Wotherspoon, A.; Salisbury, J.; Mufti, G.J.; Hamblin, T.J.; Devereux, S. CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood 2008, 111, 5173–5181. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Orchard, J.A.; Ibbotson, R.E.; Davis, Z.; Thomas, P.W.; Stevenson, F.K.; Oscier, D.G. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002, 99, 1023–1029. [Google Scholar] [CrossRef]
- Chang, C.C.; Cleveland, R.P. Conversion of CD38 and/or myeloid-associated marker expression status during the course of B-CLL: Association with a change to an aggressive clinical course. Blood 2002, 100, 1106. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, G.; Maurillo, L.; Venditti, A.; Buccisano, F.; Epiceno, A.M.; Capelli, G.; Tamburini, A.; Suppo, G.; Battaglia, A.; Del Principe, M.I.; et al. Clinical significance of CD38 expression in chronic lymphocytic leukemia. Blood 2001, 98, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Keating, M.; Do, K.A.; O’Brien, S.; Huh, Y.O.; Jilani, I.; Lerner, S.; Kantarjian, H.M.; Albitar, M. CD38 expression as an important prognostic factor in B-cell chronic lymphocytic leukemia. Blood 2001, 98, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zucchetto, A.; Vaisitti, T.; Benedetti, D.; Tissino, E.; Bertagnolo, V.; Rossi, D.; Bomben, R.; Dal Bo, M.; Del Principe, M.I.; Gorgone, A.; et al. The CD49d/CD29 complex is physically and functionally associated with CD38 in B-cell chronic lymphocytic leukemia cells. Leukemia 2012, 26, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Zucchetto, A.; Benedetti, D.; Tripodo, C.; Bomben, R.; Dal Bo, M.; Marconi, D.; Bossi, F.; Lorenzon, D.; Degan, M.; Rossi, F.M.; et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009, 69, 4001–4009. [Google Scholar] [CrossRef] [PubMed]
- Mele, S.; Devereux, S.; Pepper, A.G.; Infante, E.; Ridley, A.J. Calcium-RasGRP2-Rap1 signaling mediates CD38-induced migration of chronic lymphocytic leukemia cells. Blood Adv. 2018, 2, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft-dependent signaling complex in human B cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef]
- Krober, A.; Seiler, T.; Benner, A.; Bullinger, L.; Bruckle, E.; Lichter, P.; Dohner, H.; Stilgenbauer, S. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 2002, 100, 1410–1416. [Google Scholar] [CrossRef]
- Ottaggio, L.; Viaggi, S.; Zunino, A.; Zupo, S.; Rossi, E.; Spriano, M.; Abbondandolo, A.; Ferrarini, M. Chromosome aberrations evaluated by comparative genomic hybridization in B-cell chronic lymphocytic leukemia: Correlation with CD38 expression. Haematologica 2003, 88, 769–777. [Google Scholar]
- Smith, A.; Crouch, S.; Lax, S.; Li, J.; Painter, D.; Howell, D.; Patmore, R.; Jack, A.; Roman, E. Lymphoma incidence, survival and prevalence 2004-2014: Sub-type analyses from the UK’s Haematological Malignancy Research Network. Br. J. Cancer 2015, 112, 1575–1584. [Google Scholar] [CrossRef]
- Dreyling, M.; Campo, E.; Hermine, O.; Jerkeman, M.; Le Gouill, S.; Rule, S.; Shpilberg, O.; Walewski, J.; Ladetto, M.; Committee, E.G. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, IV62–IV71. [Google Scholar] [CrossRef]
- Espinet, B.; Ferrer, A.; Bellosillo, B.; Nonell, L.; Salar, A.; Fernandez-Rodriguez, C.; Puigdecanet, E.; Gimeno, J.; Garcia-Garcia, M.; Vela, M.C.; et al. Distinction between asymptomatic monoclonal B-cell lymphocytosis with cyclin D1 overexpression and mantle cell lymphoma: From molecular profiling to flow cytometry. Clin. Cancer Res. 2014, 20, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Galan, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L., 3rd; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Pfreundschuh, M.; Trumper, L.; Osterborg, A.; Pettengell, R.; Trneny, M.; Imrie, K.; Ma, D.; Gill, D.; Walewski, J.; Zinzani, P.L.; et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: A randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006, 7, 379–391. [Google Scholar] [CrossRef]
- Coiffier, B.; Thieblemont, C.; Van Den Neste, E.; Lepeu, G.; Plantier, I.; Castaigne, S.; Lefort, S.; Marit, G.; Macro, M.; Sebban, C.; et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: A study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 2010, 116, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Alsuwaidan, A.; Pirruccello, E.; Jaso, J.; Koduru, P.; Garcia, R.; Krueger, J.; Doucet, M.; Chaudhry, R.; Fuda, F.; Chen, W. Bright CD38 Expression by Flow Cytometric Analysis Is a Biomarker for Double/Triple Hit Lymphomas with a Moderate Sensitivity and High Specificity. Cytom. B Clin. Cytom. 2019, 96, 368–374. [Google Scholar] [CrossRef]
- Di Gaetano, R.; Gasparetto, V.; Padoan, A.; Callegari, B.; Candiotto, L.; Sanzari, M.C.; Scapinello, A.; Tagariello, G. Flow cytometry CD4(+)CD26(-)CD38(+) lymphocyte subset in the microenvironment of Hodgkin lymphoma-affected lymph nodes. Ann. Hematol. 2014, 93, 1319–1326. [Google Scholar] [CrossRef]
- Fox, C.P.; Civallero, M.; Ko, Y.H.; Manni, M.; Skrypets, T.; Pileri, S.; Kim, S.J.; Cabrera, M.E.; Shustov, A.R.; Chiattone, C.S.; et al. Survival outcomes of patients with extranodal natural-killer T-cell lymphoma: A prospective cohort study from the international T-cell Project. Lancet Haematol. 2020, 7, e284–e294. [Google Scholar] [CrossRef]
- Zaja, F.; Tabanelli, V.; Agostinelli, C.; Calleri, A.; Chiappella, A.; Varettoni, M.; Luigi Zinzani, P.; Volpetti, S.; Sabattini, E.; Fanin, R.; et al. CD38, BCL-2, PD-1, and PD-1L expression in nodal peripheral T-cell lymphoma: Possible biomarkers for novel targeted therapies? Am. J. Hematol. 2017, 92, E1–E2. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Li, P.F.; Lu, Y.; Xia, Z.J.; Huang, H.Q.; Zhang, Y.J. CD38 expression predicts poor prognosis and might be a potential therapy target in extranodal NK/T cell lymphoma, nasal type. Ann. Hematol. 2015, 94, 1381–1388. [Google Scholar] [CrossRef]
- Naik, J.; Themeli, M.; de Jong-Korlaar, R.; Ruiter, R.W.J.; Poddighe, P.J.; Yuan, H.; de Bruijn, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica 2019, 104, e100–e103. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Sriram, H.; Khanka, T.; Chatterjee, G.; Panda, D.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Patkar, N.V.; Narula, G.; et al. Flow cytometric evaluation of CD38 expression levels in the newly diagnosed T-cell acute lymphoblastic leukemia and the effect of chemotherapy on its expression in measurable residual disease, refractory disease and relapsed disease: An implication for anti-CD38 immunotherapy. J. Immunother. Cancer 2020, 8, e000630. [Google Scholar] [CrossRef]
- Farber, M.; Chen, Y.; Arnold, L.; Mollmann, M.; Boog-Whiteside, E.; Lin, Y.A.; Reinhardt, H.C.; Duhrsen, U.; Hanoun, M. Targeting CD38 in acute myeloid leukemia interferes with leukemia trafficking and induces phagocytosis. Sci. Rep. 2021, 11, 22062. [Google Scholar] [CrossRef]
- Muller, K.; Vogiatzi, F.; Winterberg, D.; Rosner, T.; Lenk, L.; Bastian, L.; Gehlert, C.L.; Autenrieb, M.P.; Bruggemann, M.; Cario, G.; et al. Combining daratumumab with CD47 blockade prolongs survival in preclinical models of pediatric T-ALL. Blood 2022, 140, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef]
- Martin, T.; Baz, R.; Benson, D.M.; Lendvai, N.; Wolf, J.; Munster, P.; Lesokhin, A.M.; Wack, C.; Charpentier, E.; Campana, F.; et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 129, 3294–3303. [Google Scholar] [CrossRef]
- van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; San-Miguel, J.; Casneuf, T.; Iida, S.; Lonial, S.; Usmani, S.Z.; Spencer, A.; Moreau, P.; Plesner, T.; Weisel, K.; et al. Evaluation of Sustained Minimal Residual Disease Negativity With Daratumumab-Combination Regimens in Relapsed and/or Refractory Multiple Myeloma: Analysis of POLLUX and CASTOR. J. Clin. Oncol. 2021, 39, 1139–1149. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Gavriatopoulou, M.; Oriol, A.; Rabin, N.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022, 23, 65–76. [Google Scholar] [CrossRef]
- Boissel, N.; Chevallier, P.; Doronin, V.; Griskevicius, L.; Maschan, A.; McCloskey, J.; Rambaldi, A.; Rossi, G.; Sokolov, A.; Wartiovaara-Kautto, U.; et al. Isatuximab monotherapy in patients with refractory T-acute lymphoblastic leukemia or T-lymphoblastic lymphoma: Phase 2 study. Cancer Med. 2022, 11, 1292–1298. [Google Scholar] [CrossRef]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef]
- Durig, J.; Naschar, M.; Schmucker, U.; Renzing-Kohler, K.; Holter, T.; Huttmann, A.; Duhrsen, U. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia 2002, 16, 30–35. [Google Scholar] [CrossRef]
- Kishimoto, H.; Hoshino, S.; Ohori, M.; Kontani, K.; Nishina, H.; Suzawa, M.; Kato, S.; Katada, T. Molecular mechanism of human CD38 gene expression by retinoic acid. Identification of retinoic acid response element in the first intron. J. Biol. Chem. 1998, 273, 15429–15434. [Google Scholar] [CrossRef]
- Ferrero, E.; Saccucci, F.; Malavasi, F. The human CD38 gene: Polymorphism, CpG island, and linkage to the CD157 (BST-1) gene. Immunogenetics 1999, 49, 597–604. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Damle, R.; Cutrona, G.; Ferrarini, M.; Chiorazzi, N. CD38 and chronic lymphocytic leukemia: A decade later. Blood 2011, 118, 3470–3478. [Google Scholar] [CrossRef]
- Prus, E.; Fibach, E. Retinoic acid induction of CD38 antigen expression on normal and leukemic human myeloid cells: Relationship with cell differentiation. Leuk. Lymphoma 2003, 44, 691–698. [Google Scholar] [CrossRef]
- Drach, J.; McQueen, T.; Engel, H.; Andreeff, M.; Robertson, K.A.; Collins, S.J.; Malavasi, F.; Mehta, K. Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res. 1994, 54, 1746–1752. [Google Scholar]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Minnema, M.C.; Levin, M.D.; Broijl, A.; Bos, G.M.J.; Kersten, M.J.; Mutis, T.; Verkleij, C.P.M.; Nijhof, I.S.; Maas-Bosman, P.W.C.; et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5128–5139. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Wu, X.; Chu, S.; Wang, J.; Yuan, H.; Roth, M.; Yuan, Y.C.; Bhatia, R.; Chen, W. ATRA-induced cellular differentiation and CD38 expression inhibits acquisition of BCR-ABL mutations for CML acquired resistance. PLoS Genet. 2014, 10, e1004414. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Perez-Simon, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Atadja, P.; Pandiella, A.; San Miguel, J.F. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006, 66, 5781–5789. [Google Scholar] [CrossRef]
- Sanchez, E.; Shen, J.; Steinberg, J.; Li, M.; Wang, C.; Bonavida, B.; Chen, H.; Li, Z.W.; Berenson, J.R. The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk. Res. 2011, 35, 373–379. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Gotz, R.; Doose, S.; Sauer, M.; Rodriguez-Gil, A.; Nerreter, T.; Kortum, K.M.; Perez-Simon, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef]
- Wang, H.F.; Ning, F.; Liu, Z.C.; Wu, L.; Li, Z.Q.; Qi, Y.F.; Zhang, G.; Wang, H.S.; Cai, S.H.; Du, J. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. 2017, 66, 355–366. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Endell, J.; Boxhammer, R.; Wurzenberger, C.; Ness, D.; Steidl, S. The Activity of MOR202, a Fully Human Anti-CD38 Antibody, Is Complemented by ADCP and Is Synergistically Enhanced by Lenalidomide In Vitro and In Vivo. Blood 2012, 120, 4018. [Google Scholar] [CrossRef]
- Endell, J.; Boxhammer, R.; Steidl, S. Synergistic in Vitro Activity of MOR202, a Human CD38 Antibody, in Combination with Pomalidomide. Blood 2014, 124, 5712. [Google Scholar] [CrossRef]
- Boxhammer, R.; Steidl, S.; Endell, J. Effect of IMiD compounds on CD38 expression on multiple myeloma cells: MOR202, a human CD38 antibody in combination with pomalidomide. J. Clin. Oncol. 2015, 33, 8588. [Google Scholar] [CrossRef]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.N.; Huntington, N.D.; Shi, W.; Huang, D.C.S.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Liu, H.; Fang, C.; Li, C.; Xhyliu, F.; Dysert, H.; Bodo, J.; Habermehl, G.; Russell, B.E.; Li, W.; et al. Targeting of CD38 by the Tumor Suppressor miR-26a Serves as a Novel Potential Therapeutic Agent in Multiple Myeloma. Cancer Res. 2020, 80, 2031–2044. [Google Scholar] [CrossRef]
- Jude, J.A.; Dileepan, M.; Subramanian, S.; Solway, J.; Panettieri, R.A., Jr.; Walseth, T.F.; Kannan, M.S. miR-140-3p regulation of TNF-alpha-induced CD38 expression in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L460–L468. [Google Scholar] [CrossRef]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Ferracin, M.; Ghimenti, C.; Massaia, M.; Horenstein, A.L.; Malavasi, F. Anti-CD38 antibody therapy: Windows of opportunity yielded by the functional characteristics of the target molecule. Mol. Med. 2013, 19, 99–108. [Google Scholar] [CrossRef]
- Funaro, A.; Reinis, M.; Trubiani, O.; Santi, S.; Di Primio, R.; Malavasi, F. CD38 functions are regulated through an internalization step. J. Immunol. 1998, 160, 2238–2247. [Google Scholar]
- Taylor, R.P.; Lindorfer, M.A. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef]
- Chillemi, A.; Quarona, V.; Zito, A.; Morandi, F.; Marimpietri, D.; Cuccioloni, M.; Robert, O.J.; Mark, C.S.; Bolzoni, M.; Toscani, D.; et al. Generation and Characterization of Microvesicles after Daratumumab Interaction with Myeloma Cells. Blood 2015, 126, 1849. [Google Scholar] [CrossRef]
- Malavasi, F.; Faini, A.C.; Morandi, F.; Castella, B.; Incarnato, D.; Oliviero, S.; Horenstein, A.L.; Massaia, M.; van de Donk, N.; Richardson, P.G. Molecular dynamics of targeting CD38 in multiple myeloma. Br. J. Haematol. 2021, 193, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, N.; Azaman, M.I.; Chng, W.J. Daratumumab Resistant Natural Killer/T-Cell Lymphoma Exhibit an Addiction to the Exosome Biogenesis Pathway for Survival. Blood 2021, 138, 2256. [Google Scholar] [CrossRef]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef]
- van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef]
- Kortum, K.M.; Zhu, Y.X.; Shi, C.X.; Jedlowski, P.; Stewart, A.K. Cereblon binding molecules in multiple myeloma. Blood Rev. 2015, 29, 329–334. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Kastritis, E.; Ntanasis-Stathopoulos, I.; Fotiou, D.; Roussou, M.; Migkou, M.; Ziogas, D.C.; Kanellias, N.; Terpos, E.; Dimopoulos, M.A. The addition of IMiDs for patients with daratumumab-refractory multiple myeloma can overcome refractoriness to both agents. Blood 2018, 131, 464–467. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Jhatakia, A.; Broekmans, M.E.C.; Frerichs, K.A.; Zweegman, S.; Mutis, T.; Bezman, N.A.; van de Donk, N. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers 2020, 12, 3713. [Google Scholar] [CrossRef]
- Wang, X.; Yu, X.; Li, W.; Neeli, P.; Liu, M.; Li, L.; Zhang, M.; Fang, X.; Young, K.H.; Li, Y. Expanding anti-CD38 immunotherapy for lymphoid malignancies. J. Exp. Clin. Cancer Res. 2022, 41, 210. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 84. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, X.; Li, X.; Zhou, Y.; Song, Z.; Zhang, J.; Shi, B.; Wang, J. Novel BCMA-OR-CD38 tandem-dual chimeric antigen receptor T cells robustly control multiple myeloma. Oncoimmunology 2021, 10, 1959102. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-directed CAR-T cell therapy: A novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef]
- Fayon, M.; Martinez-Cingolani, C.; Abecassis, A.; Roders, N.; Nelson, E.; Choisy, C.; Talbot, A.; Bensussan, A.; Fermand, J.P.; Arnulf, B.; et al. Bi38-3 is a novel CD38/CD3 bispecific T-cell engager with low toxicity for the treatment of multiple myeloma. Haematologica 2021, 106, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggio, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.; Nijhof, I.S. Resistance Mechanisms Towards CD38-Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Nagai, Y.; Elmas, E.; de Souza Fernandes Pereira, M.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]
- You, T.; Hu, W.; Ge, X.; Shen, J.; Qin, X. Application of a novel inhibitor of human CD59 for the enhancement of complement-dependent cytolysis on cancer cells. Cell Mol. Immunol. 2011, 8, 157–163. [Google Scholar] [CrossRef]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; De Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafa, N.; Azaman, M.I.; Ng, G.G.K.; Chng, W.J. Molecular Determinants Underlying the Anti-Cancer Efficacy of CD38 Monoclonal Antibodies in Hematological Malignancies. Biomolecules 2022, 12, 1261. https://doi.org/10.3390/biom12091261
Mustafa N, Azaman MI, Ng GGK, Chng WJ. Molecular Determinants Underlying the Anti-Cancer Efficacy of CD38 Monoclonal Antibodies in Hematological Malignancies. Biomolecules. 2022; 12(9):1261. https://doi.org/10.3390/biom12091261
Chicago/Turabian StyleMustafa, Nurulhuda, Muhamad Irfan Azaman, Giselle G. K. Ng, and Wee Joo Chng. 2022. "Molecular Determinants Underlying the Anti-Cancer Efficacy of CD38 Monoclonal Antibodies in Hematological Malignancies" Biomolecules 12, no. 9: 1261. https://doi.org/10.3390/biom12091261
APA StyleMustafa, N., Azaman, M. I., Ng, G. G. K., & Chng, W. J. (2022). Molecular Determinants Underlying the Anti-Cancer Efficacy of CD38 Monoclonal Antibodies in Hematological Malignancies. Biomolecules, 12(9), 1261. https://doi.org/10.3390/biom12091261