1. Introduction
Biological lipids are a heterogeneous group of non-polar compounds that perform a range of critical functions from compartmentalization to energy storage to signaling [
1]. Unlike genes and proteins, biological lipid synthesis is not template driven; the dynamic lipidome is an interaction between lipid metabolic enzyme activity and substrate accessibility which is influenced by the environment (including diet). Biological lipids are classified by their chemical structure into eight classes based on the hydrophobic and hydrophilic components of the lipid [
2]. However, the structure–function relationship of lipids goes beyond the class, with features such as fatty acid chain length, number and position of saturation (double bond) influencing lipid function [
3]. Furthermore, the relative composition of lipid classes modifies the physical properties of membranes [
3]. Therefore, the goals of lipidomics are to profile the composition of the lipidome and to detect and quantify lipid species. While advances in mass spectrometry technology and development of analytical and lipid-specific informatics methods have enabled high-throughput lipidomics, a validated method of sample quality control is yet to be established.
In principle, the steps in the workflow for lipidomics should parallel the other, more developed omics, and comprise sample extraction, quality control, and total lipid quantification steps prior to normalization of lipid sample amount for molecular profiling. However, sample quality control and normalization of loading by the total lipid amount in a sample is currently absent from the recommendations for good practice in lipidomics [
4], possibly due to the lack of validated, fit-for-purpose, sample-preserving methods for lipid sample quality control prior to mass spectrometry analysis. Without total lipid quantification, sample volume (e.g., serum/plasma) or weight (e.g., tissue) have been used as the basis of normalization. While this approach is appropriate for studies using well-characterized sample types in cross-sectional or well-controlled studies, a sample quality control and quantification step is essential for novel, less-controlled sample types; for example, materials derived from foodstuffs, plant samples and wildlife.
Despite the heterogeneity of biological lipids, we reasoned that total lipid quantity and quality assessment may be accomplished through spectroscopic measurements of common lipid structures such as hydrocarbons and ester bonds, which are generally absent in contaminating biochemicals. A similar application is used in the RNA sequencing workflow, where preparations of RNA from any source are routinely assessed for quality and quantity via UV/VIS spectroscopy (NanoDrop spectrometers) using a generalized metric of RNA-specific peaks (absorbance at 260 nm, A260) despite different absorbance peaks for adenosine, uridine (260 nm), guanosine (254 nm), and cytosine (271 nm) [
5]. Ratios between A260 and absorbance of known interfering molecules (A230, A280) are used to assess sample purity [
6].
For lipid quantification, there is an existing commercial product (DirectDetect
TM) [
7] which uses Fourier-transform infrared (FTIR) spectroscopy with an external standard to quantify lipids based on CH symmetric stretching vibration between 2870 and 2840 cm
−1 [
8]. While this simple formula is accurate for pure lipids, the quantification accuracy of complex lipid samples/mixtures, and the potential impact of different lipid extraction methods and biochemical contamination on total lipid quantification by FTIR have not yet been fully assessed. Since the presence of other (bio)chemicals in the sample can influence IR absorbance for CH stretching due to the masking effect [
9], and lipidomics experiments almost always analyze complex (rather than pure) lipids, a technical assessment and validation of complex lipid quantification by FTIR are essential prior to adoption into the workflow.
Here, we conducted a systematic evaluation of the parameters that impact total complex lipid mixture quantification by FTIR and developed new formulae to facilitate the use of FTIR for lipid sample quality assessment. We chose the Attenuate Total Reflectance (ATR) sampling method for FTIR because it requires just one microliter of MS-ready sample without additional sample preparation. The organic solvent from the lipid extracts readily evaporates on the ATR sensor, allowing rapid FTIR data acquisition. Firstly, we examined the impact of sample extraction method and FTIR spectra local baseline correction on the ensuing lipid quantification against the external lipid standard curve using either a single lipid or a lipid mixture. For this assessment, we compared FTIR spectra of a cellular lipid extract using commonly used monophasic and biphasic extraction methods to pure lipid standard and the mixed cell extract (with all biomolecules). Secondly, we assessed common biochemical contaminants that may occur during sample preparation and lipid extraction (e.g., incomplete metabolite removal, trace sucrose, detergents), or due to contaminants in the solvent or tubes (e.g., PEG). We titrated a range of common biochemical contaminants to a pure lipid extract to assess the sensitivity ATR-FTIR for contaminant detection. Based on the results of these extensive investigations, we developed a lipid quality (LiQ) score to detect contamination of lipid samples. Finally, the LiQ score was evaluated on a set of plasma samples with comparison to LC-MS-based lipidomics.
2. Materials and Methods
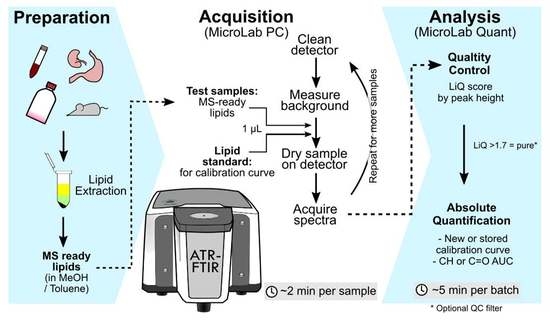
Figure 1 depicts the experimental design formulated to assess the parameters for using ATR-FTIR spectroscopy for quality and quantity assessment of complex lipid mixtures, which are the starting materials for lipidomics.
2.1. Materials
Stearic acid (S4751), sucrose (84097), glucose (G8270), galactose (G0750), NP-40 (542334), Triton-X100 (T8787), sodium deoxycholate (D6750), 1-butanol (34867), 2,6-di-tert-butyl-4-methyl-phenol (BHT, B1378), tert-butyl-methyl-ether (TBME, 34875), 2-aminoanthracene (A38800) and Val-Tyr-Val (V8376) were purchased from Sigma Aldrich (Castle Hill, Australia). RNA primer (Qiagen, MS00003556) was purchased from Qiagen (Clayton, Australia). SPLASH LipidoMix (330707) was purchased from Avanti Polar Lipids, inc. (Alabaster, AL, USA). Agilent API-TOF Reference Mix (G1969-85001), 250 µL PP inserts with graduation (#5190-4073), and PTFE/silicone rubber septa (#5182-0731) were ordered from Agilent Technologies (Mulgrave, VIC, Australia). Methanol (A456-4), 2-propanol (A451-4), and acetonitrile (A955-4) were purchased from Thermo Fisher Scientific (Scoresby, Australia). The Synergy UV Water Purification System was used to filter MilliQ water (Merck Millipore, Burlington, MA, USA).
2.2. Cell Culture and Lipid Extract Preparation
PC3 (RRID: CVCL_0035) cells were grown in 5% FBS (Bovogen, SFBS-FR)/RPMI-1460 (Gibco, 11875119) culture medium at 37 °C, 5% CO2. Cells were transferred into 10 cm Petri dishes, harvested by cell scraping, and counted using a hemocytometer. To prepare three extracts with varying levels of contamination, aliquots of one million cells were prepared, pelleted, and washed with PBS, and then processed using one of the following methods. For the “Mixed sample” of all cellular components, the cell pellet was resuspended with 300 µL butanol:methanol, followed by immediate analysis of the cell extract by FTIR spectroscopy. In case of the “BuMe” extract, the cell pellet was resuspended with 300 µL butanol:methanol, then incubated overnight at −20 °C. Protein was then removed by centrifugation at 16,000× g for 30 min at 4 °C, and the supernatant was collected in a separate tube and analyzed using FTIR spectroscopy. For the “TBME” extract, the cell pellet was resuspended in 200 µL of chilled methanol, sonicated for 1 min, and incubated overnight at −20 °C. TBME (tert-butyl-methyl-ether, 700 µL) was added and the sample was vortexed before adding 180 µL of milliQ water. After mixing, the sample was centrifuged at 16,000× g for 30 min at 4 °C and the upper (lipid extract) and middle (metabolite extract, for quality control analysis) phases were collected into individual tubes. The lipid extract was then dried, resuspended in 300 µL butanol:methanol, and analyzed via FTIR spectroscopy. The remaining protein pellet was washed and resuspended (50 µL) in PBS, and the amount of protein was quantified by BCA assay for use in quality control analyses.
2.3. Preparation of Contaminant Samples
Stock mixtures of each contaminant were generated via the following methods. Solid sucrose, glucose, and galactose were resuspended in 10 µL of MilliQ water before dilution in 3:1 butanol:methanol to a final volume of 1 mL and concentration of 1 µg/µL. Protein extracted from cell lysates was diluted in 3:1 butanol:methanol to provide a 1 mL stock solution with a protein concentration of 1 µg/µL. Due to the precipitation of protein and sugars in organic solvents, these samples were thoroughly vortexed before each use, measurement, or dilution. Solid detergent (sodium deoxycholate) were resuspended in 3:1 butanol:methanol to a concentration of 1% (w/v), whereas liquid detergent (Triton X-100, NP-40) was diluted to 1% (v/v) in 3:1 butanol:methanol. Lyophilised RNA primer was resuspended in 10 µL MilliQ water and diluted to 1 µg/µL (200 µL) in 3:1 butanol:methanol, and its concentration confirmed using a NanoDrop 2000/2000c spectrometer (Thermo Fisher Scientific). Metabolites extracted from 107 cells (method described above) were collected from the aqueous phase of a TBME extraction (380 µL), dried, and resuspended in 380 µL 3:1 butanol:methanol. Lipids purified by TBME extraction were then quantified by ATR-FTIR spectroscopy and diluted to 1 µg/µL for use as pure lipid diluent for quality-control analysis. Lipids extracted using the BuMe method were diluted using the same dilution factor as TBME lipids.
Dilutions for quality control analysis were prepared by spiking variable amounts of contaminant stock into 10 µL pure lipid extract, followed by drying and resuspension in 10 µL 3:1 butanol:methanol. This ensures a constant lipid concentration of 1 µg/µL without dilution from the contaminant that has been spiked in.
2.4. Human Plasma Sample Collection
Deidentified plasma samples from a cohort of patients with chronic liver disease were used for proof-of-concept application of lipid extract quality assessment. These samples were collected following informed consent from the participants. The work was approved by the research ethics committees of the Prince Charles Hospital (HREC/15/QPCH/202) and QIMR Berghofer Medical Research Institute (P2352), and abided by the Declaration of Helsinki principles. Blood was collected using EDTA as the anticoagulant. Following centrifugation of the samples, plasma was collected and stored at −80 °C.
2.5. Human Plasma Sample Preparation
Plasma samples were thawed on ice prior to metabolite and lipid extractions. All pipetting steps were performed in a cold room or on ice. Plasma (30 µL) was mixed with 270 µL of butanol/methanol (1:1 v/v) containing 10 mM ammonium formate, 50 µg/mL BHT, and 1.5 µL SPLASH internal standard mixture. Samples were agitated using a thermomixer and sonicated (25 °C, 850 rpm, 1 h). Samples were centrifuged at 16,000× g (20 °C, 15 min) before aliquoting 100 µL of the supernatant for mass spectrometry analysis. For FTIR spectra acquisition, 100 µL of butanol:methanol lipid extracts were aliquoted into a Greiner V-bottom 96-well plate and dried using an evaporative sample concentrator (Genevac EZ-2, Marshall scientific). The samples were reconstituted in 12 µL of butanol:methanol (1:1).
2.6. ATR-FTIR Spectroscopy
An Agilent Cary 630 fitted with an ATR module was used for the acquisition of spectra. Mid-infrared spectra (4000–650 cm−1) were collected at a resolution of 8 cm−1 using 64 scans per acquisition. The detector stage was cleaned with 80% ethanol, and the background spectra (ambient room air at 21 °C) were collected. A lipid sample (1 μL) or standard in butanol:methanol mixture was applied to the detector and allowed to air dry (~30 s). Lipid standards were prepared by serial dilution of stearic acid and Avanti SPLASH LipidoMix in butanol:methanol. All spectra were baseline corrected using the baseline algorithm built into the Agilent MicroLab Expert software with set regions 2031–1865 cm−1 and 3971–3799 cm−1. Note that the baseline correction adjusts the entire spectra baseline to the experimental baseline (blank), which is different to the local baseline subtraction procedure that we test during this study. Local baseline subtraction was not applied to all spectra. The spectra were then exported from MicroLab Expert (Agilent) software as CSV files.
2.7. LC-MS Lipidomics
Targeted lipidomics experiments were performed following previously published methodologies [
10]. Briefly, an Agilent Technologies 1290 Infinity II UHPLC system with an Agilent ZORBAX eclipse plus C18 column (2.1 × 100 mm 1.8 µm) (#959758-902) and guard column (#821725-901), coupled online with an Agilent 6470 triple quadrupole system, was used for the targeted lipidomics experiments. The instrument was tuned in positive ionization mode and unit resolution. Buffer A contained 10 mM ammonium formate in water/acetonitrile/isopropanol (50:30:20%
v/
v/
v), whereas buffer B contained 10 mM ammonium formate in water/acetonitrile/isopropanol (1:9:90%
v/
v/
v). A multi-wash procedure was performed prior to each sample injection. In this procedure, the needle was washed and needle seat back flushed with isopropanol, MilliQ water, and acetonitrile to reduce sample carryover.
The source nitrogen gas temperature was set to 175 °C at a flow of 11 L/min and a sheath gas temperature of 250 °C at a flow of 10 L/min. The capillary voltage was set to 3500 V and nozzle voltage to 0 V for positive mode, and the nebulizer operated at 20 psi. Check tunes were performed in wide, unit, and enhanced modes prior to each experiment to confirm the performance of the mass spectrometer. The quadrupole was tuned to reference masses 118.09, 322.05, 622.03, 922.01, and 1221.99 in positive ionization mode; 112.99, 302.00, 601.98, 1033.99, and 1333.97 in negative ionization mode.
The instrument was operated in dynamic MRM mode using the transitions published by Huynh et al. [
10], including LipidoMix internal standards. Six microliters of sample were injected per acquisition. Acquired data were imported into Skyline (MacCoss Lab, Department of Genome Sciences, University of Washington, Seattle, DC, USA) [
11]. Peak integration was automated, but it was manually confirmed and corrected if required. Internal standard retention time was used to confirm correct peak integration of lipids belonging to the same class. Peak areas were exported from Skyline for further analysis in R (R Foundation for Statistical Computing, Vienna, Austria) [
12]. Total intensity (TI) values were generated by summing all lipid species intensities per acquisition and adjusted to reflect differences in concentration between MS measurements (6 µL of 100 µL plasma lipid sample, 6% total sample measured per acquisition) and FTIR spectroscopy (1 µL of concentrated plasma lipid sample, 8.33% total sample measured per acquisition).
2.8. Data Analysis and Statistics
Method development and optimization analysis were completed using R (version 4.0.2) and Graphpad Prism software, where the final application utilized MicroLab Quant and Expert software. Baselined spectra were analyzed using R studio and the DescTools package (0.99.44). All analyses were completed on baselined spectra to standardize all spectra to the same baseline. No normalization was completed on these spectra due to the quantitative nature of the analysis. The area under the curve (AUC) was calculated for each desired region using the trapezoid method via the AUC function. For analysis in
Figure 2C only, a local baseline was calculated using the minimum and maximum range values in the trapezoid area formula [area = (Height
Min−Height
Max)/2 × (Wavenumber
Max−Wavenumber
Min)] and subtracted from each AUC to result in a locally baselined AUC value. Mean baselined AUC and standard deviation were calculated across the technical replicates (
n = 3−6) for each sample. Simple linear regression analysis and graphs were generated by Graphpad Prism (v8.4.3., GraphPad Software Inc., San Diego, CA, USA) for each standard lipid calibration curve. Signal to noise ratios (S/N), limits of detection (LOD) and quantitation (LOQ) were calculated using the International Conference on Harmonization method [
13]. FTIR and mass spectrometry measures were compared by Pearson correlation (R value) and simple linear regression analysis (R
2 and
p values). Two-sided two-way ANOVA test comparing analysis parameters was conducted using GraphPad Prism, with Sidak’s multiple comparisons. Graph collation and diagrams were generated by Inkscape.
4. Discussion
Here, we present a comprehensive evaluation of ATR-FTIR for complex lipid extract quantification and propose an integrated, simple, sample-conserving method to assist in lipidomics sample quality assessment prior to mass spectrometry analysis. There are several important reasons to evaluate lipid sample quality early in the lipidomics workflow. Firstly, certain contaminants, such as detergents, can confound MS data and even impact LC-MS performance for later samples in the sequence. Early detection of contaminated samples will allow elimination of these sample from the analysis, or re-extraction of the sample. Secondly, contamination can also confound total lipid quantification by FTIR, as illustrated in
Figure 3 and applied in
Figure 4. While we have devised a simple LiQ Score to facilitate screening for exclusion of low-quality samples, closer analysis and interpretation of spectral features can provide additional information on the potential contaminants in these samples. To this end, we have offered three follow-on ratiometric analyses in
Figure S1. These additional peak height ratios compare the contribution of lipid peak versus detergent, protein, or sugar peaks individually to clarify the composition of the sample and potentially inform the source of contaminants.
Our evaluation of lipid quantification by FTIR highlights a detrimental impact of baseline subtraction on the CH region AUC, which we determined to be more accurate than the alternative C=O peak. Baseline subtraction is also referred to as baseline correction, or anchoring, and can be a default parameter in FTIR spectral processing software [
7,
14]. While useful in some applications, quantification of peaks in complex mixtures with overlapping spectra faces augmentation of the local baseline absorbance. This bisects the target peak and results in partial exclusion of peak areas that are masked by the non-target peaks, leading to underestimates in terms of quantity. More sophisticated methods such as spectral deconvolution could be employed to accurately separate the target from non-target peaks, as demonstrated in inorganic chemistry applications [
25]. However, attempts for complex biological features have been met with difficulty [
21]. We caution users about the use of local baseline correction for FTIR AUC-based lipid quantification calculations, especially in instances of non-pure lipid samples.
Our detailed assessment of the calibration curve of a simple lipid (stearic acid) and a complex (SPLASH LipidoMix) lipid mixture showed a similar signal-to-noise ratio, linearity, limit of detection, and quantification. SPLASH LipidoMix is produced to reflect the human plasma composition, comprising of physiologically accurate concentrations of phospholipids, sphingolipids, cholesterol, cholesterol esters, and glycerides, whereas stearic acid merely reflects a simple fatty acid of 18-carbon length. The difference in lipid composition of the two standards is evident in the full spectra (
Figure 2A), where amine, phosphodiester, and sugar groups are present in the complex mixture reflecting an expected biological sample. Prediction of lipid quantities was not impacted by this added complexity. Instead, it was highly dependent on the hydrocarbon chain length and number of chains (bound to head group by the lipid ester group) of the chosen lipid standards and sample. Use of a simple lipid standard should be chosen to reflect the average lipid chain and number of the sample for the closest accuracy due to this relationship. Other FTIR-based analyses have been proposed to delineate lipid class composition and shifts in lipid spectra in disease [
19,
26], whereby use of different species of lipid as standard may also provide some frame of reference for specific lipid classes or disease information.
Compared to the commercial product DirectDetect, which uses transmission mode for spectra acquisition, there are several advantages of our proposed ATR-FTIR lipid quality and quantity assessment workflow. DirectDetect outputs the calculated lipid quantity using the AUC from the CH region 2870–2840 cm
−1 with baseline anchoring, which is narrower than our CH band (3000–2800 cm
−1), and the recommendation to not baseline subtract. As far as we are aware, DirectDetect does not allow the opportunity to modify the quantification method or ranges to customize use [
27]. The stated limit of detection for DirectDetect is 250 ng/μL, which is ten-fold higher than the proposed ATR-FTIR workflow [
7]. No additional consumables are required in our workflow, although analysis is carried out one sample at a time. The DirectDetect sample cards hold four samples at a time, but it takes several minutes to read each card [
27]. Our workflow requires approximately 2 min for each sample. While 1 µL of lipid sample was demonstrated as feasible in the ATR-FTIR workflow as an effort to minimize sample wastage, pipetting of small volumes of organic solvent can be difficult. However, analysis of the ATR-FTIR technical replicates from the 107 plasma samples showed coefficient of variance of <5% (
Figure S2), indicative of high reproducibility. For the best accuracy, we recommend using air-displacement pipettes, keeping samples at a low temperature during workflow, and conditioning the pipette tip by aspirating and dispensing the sample at least twice before transferring sample to the detector. Technical replicates may also be advised to gauge pipetting accuracy.
One of the benefits of the ATR-FTIR workflow is the customization of the parameters for other applications. Overall, this analysis relies on generalization of lipid structure, using only the hydrocarbon CH and ester C=O regions as proxy for lipid content, which allows this method to be used broadly. This is similar to UV/VIS spectroscopy for RNA analysis using only the A260 measure for nucleic acid despite variation in spectral peaks for each nucleotide [
5]. Here, we assessed the CH and C=O region as two possible generalized ranges for lipid quantification based on their prevalence in complex biological samples for lipidomics experiments. However, some applications and sample sources may require adjustment of these parameters. While lipidomics samples for mass spectrometry are prepared in organic solvent, other applications may find lipid in aqueous suspensions and thus may be more suitable for using the C=O region for quantification due to the overlapping water signal hindering CH region detection [
28]. Unsaturation of lipids results in an additional CH peak between 3010–3000 cm
−1 and a reduction in the CH-region (3000–2800 cm
−1,
Figure S3). While in human-derived samples, the unsaturation signal is minor compared to saturated signals, samples rich in unsaturated lipid may require an extension of the CH region to encompass this 3010 cm
-1 peak. This analysis of CH peaks may be extended as a method for measuring lipid unsaturation in the future. Additionally, fatty acids contain a carboxyl peak (~1710 cm
−1) as opposed to a lipid ester peak (~1740 cm
−1), and therefore extension of the C=O region may be beneficial for highly fatty-acid-enriched samples.




 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}