

Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution

 , , and
, , and 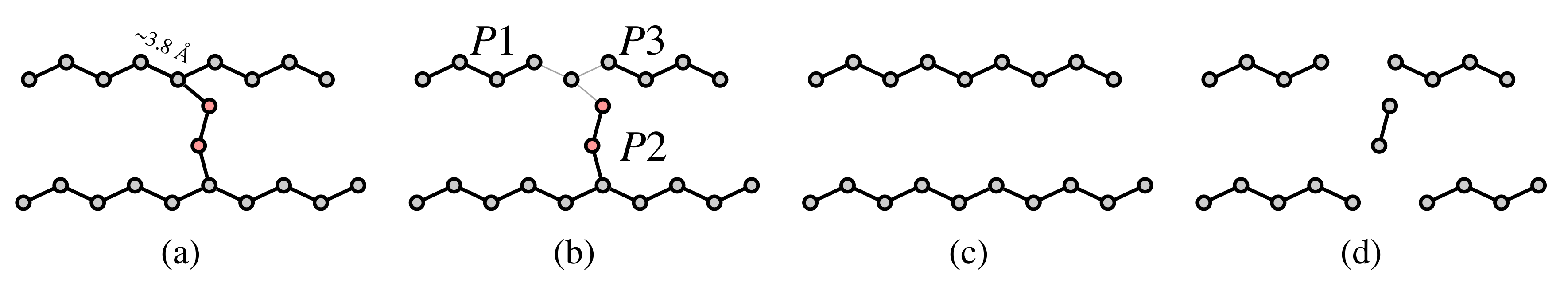{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- At resolution better than 3.3 Å the amino acid residue side-chains are visible. This information can be used to unambiguously identify the amino acids and, therefore, to trace the protein sequence into the EM density.
- From 3.3 to 4.5 Å resolution, side-chains are only partially visible. For this “shadow” range, topological reconstruction of the protein is often tedious unless complementary information is available.
- For resolution worse than 4.5 Å only some secondary structures—mainly helices—are visible. The -strands are no longer separated, rendering the topological reconstruction somewhat impossible.
2. Materials and Methods
2.1. Tree Representation of an EM Map and Minimum Spanning Tree
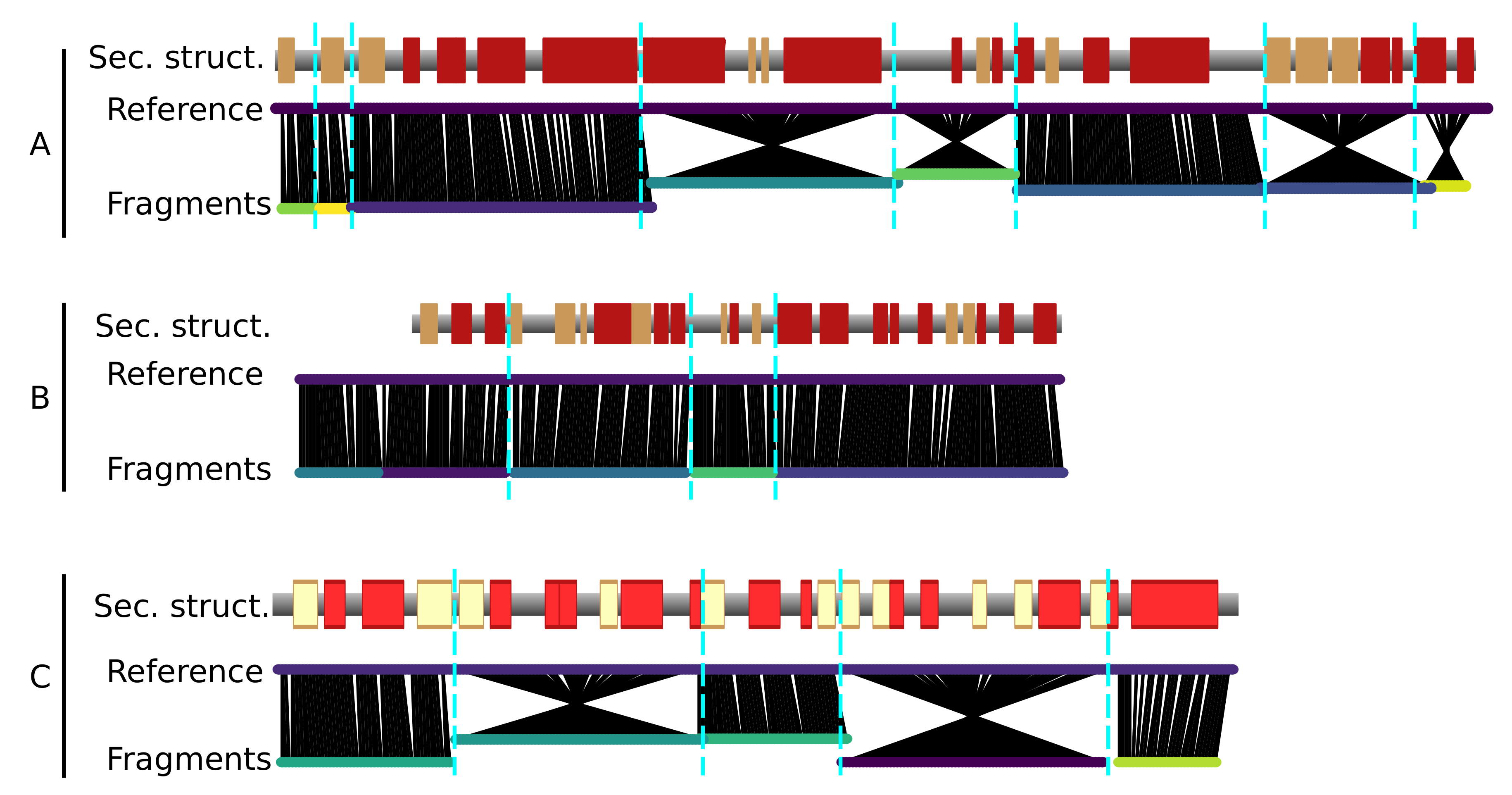
2.2. Fragment Assembly and Map Segmentation
2.3. Building the Full Atomic Model
3. Results
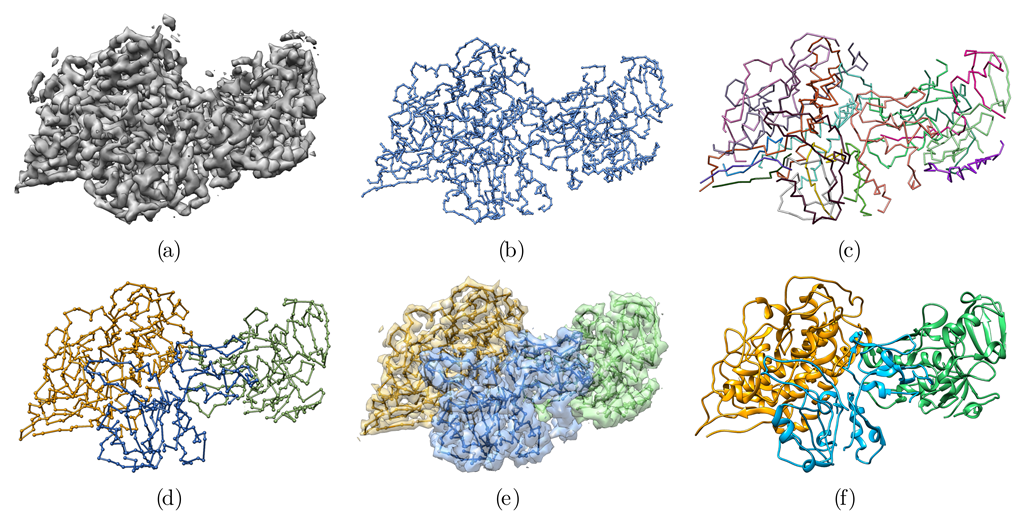
3.1. Method Principle
- (1)
- The cryo-EM map is initially represented as a weighted graph and simplified into a minimum spanning tree (Figure 2b) (MST). This tree is pruned until the maximal degree of the graph (i.e., number of edges of the vertex with the greatest number of edges incident to it) is three.
- (2)
- The tree is further pruned to remove forks in the tree, and to produce potential fragments of the polypeptide chain. At this stage, the fragments have an arbitrary N-to-C terminal orientation.
- (3)
- The fragments are concatenated by making use of evolutionary information in the form of predicted inter-residue contacts. We used the Raptorx ContactMap server [25] to predict the contacts. The direction of each fragment is determined to maximize the agreement with the predicted contacts. During this stage, the map is segmented automatically, as a consequence of the fragment concatenation procedure. No prior segmentation of the map to individual components is necessary.
- (4)
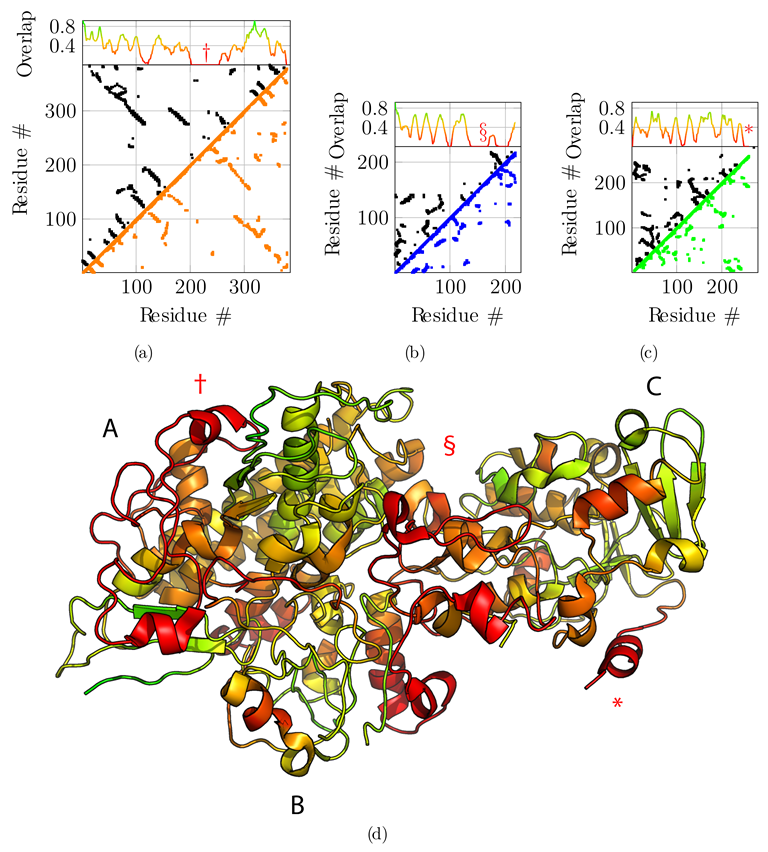
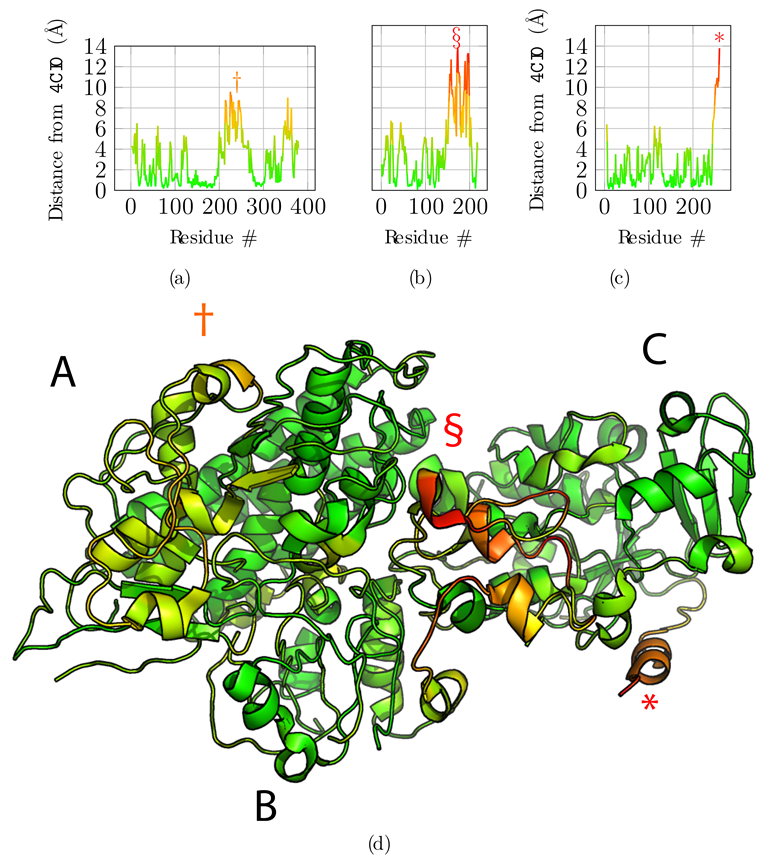
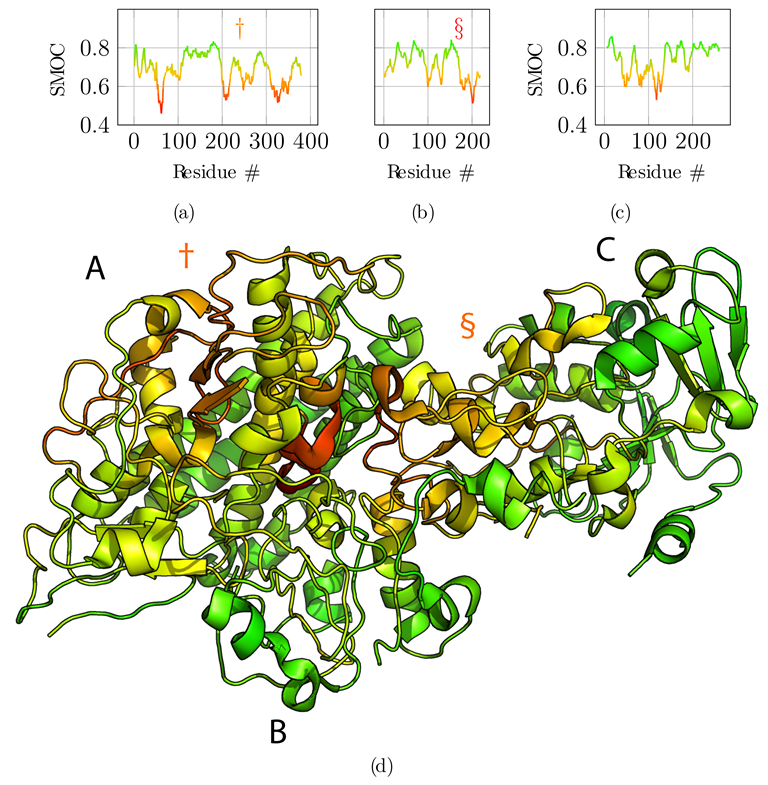
3.2. Application to the F420-Reducing Hydrogenase Heterotrimer
3.3. Application to the T6SS Baseplate with Manual Tracing
3.4. Additional Challenging Targets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radermacher, M.; Wagenknecht, T.; Verschoor, A.; Frank, J. Three-dimensional structure of the large ribosomal subunit from Escherichia coli. Embo J. 1987, 6, 1107–1114. [Google Scholar] [CrossRef]
- DiMaio, F.; Chiu, W. Tools for Model Building and Optimization into Near-Atomic Resolution Electron Cryo-Microscopy Density Maps. Methods Enzymol. 2016, 579, 255–276. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-particle cryo-EM at atomic resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Kucukelbir, A.; Sigworth, F.J.; Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 2014, 11, 63–65. [Google Scholar] [CrossRef]
- Allen, G.S.; Stokes, D.L. Modeling, docking, and fitting of atomic structures to 3D maps from cryo-electron microscopy. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2013; Volume 955, pp. 229–241. [Google Scholar] [CrossRef]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef]
- Lamzin, V.S.; Perrakis, A.; Wilson, K.S. ARP/wARP—Automated model building and refinement. Crystallogr. Biol. Macromol. 2012, F, 525–528. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Terashi, G.; Kihara, D. De novo main-chain modeling for em maps using MAINMAST. Nat. Commun. 2018, 9, 1618. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Adams, P.D.; Afonine, P.V.; Sobolev, O.V. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat. Methods 2018, 15, 905–908. [Google Scholar] [CrossRef]
- Wang, R.Y.R.; Kudryashev, M.; Li, X.; Egelman, E.H.; Basler, M.; Cheng, Y.; Baker, D.; DiMaio, F. De novo protein structure determination from near-atomic resolution cryo-EM maps. Nat. Methods 2015, 12, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Pfab, J.; Phan, N.M.; Si, D. DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl. Acad. Sci. USA 2021, 118, e2017525118. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lin, P.; Chen, J.; Cao, H.; Huang, S.Y. Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly. Nat. Commun. 2022, 13, 4066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, B.; Freddolino, P.L.; Zhang, Y. CR-I-TASSER: Assemble protein structures from cryo-EM density maps using deep convolutional neural networks. Nat. Methods 2022, 19, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Cherrak, Y.; Rapisarda, C.; Pellarin, R.; Bouvier, G.; Bardiaux, B.; Allain, F.; Malosse, C.; Rey, M.; Chamot-Rooke, J.; Cascales, E.; et al. Biogenesis and structure of a type VI secretion baseplate. Nat. Microbiol. 2018, 3, 1404–1416. [Google Scholar] [CrossRef]
- Pintilie, G.; Chiu, W. Comparison of Segger and other methods for segmentation and rigid-body docking of molecular components in Cryo-EM density maps. Biopolymers 2012, 97, 742–760. [Google Scholar] [CrossRef]
- Zhou, A.; Rohou, A.; Schep, D.G.; Bason, J.V.; Montgomery, M.G.; Walker, J.E.; Grigorieffniko, N.; Rubinstein, J.L. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. eLife 2015, 4, e10180. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007, 157, 281–287. [Google Scholar] [CrossRef]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Ovchinnikov, S.; Park, H.; Varghese, N.; Huang, P.S.; Pavlopoulos, G.A.; Kim, D.E.; Kamisetty, H.; Kyrpides, N.C.; Baker, D. Protein structure determination using metagenome sequence data. Science 2017, 355, 294–298. [Google Scholar] [CrossRef]
- Schaarschmidt, J.; Monastyrskyy, B.; Kryshtafovych, A.; Bonvin, A. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins 2018, 86 (Suppl. S1), 51–66. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Headd, J.J.; Terwilliger, T.C.; Adams, P.D. New tool: Phenix real space refine. Comput. Crystallogr. Newsl. 2013, 4, 43–44. [Google Scholar]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef]
- Allegretti, M.; Mills, D.J.; McMullan, G.; Kühlbrandt, W.; Vonck, J. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife 2014, 3, e01963. [Google Scholar] [CrossRef]
- Mills, D.J.; Vitt, S.; Strauss, M.; Shima, S.; Vonck, J. De novo modeling of the F(420)-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. eLife 2013, 2, e00218. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef]
- Joseph, A.P.; Malhotra, S.; Burnley, T.; Wood, C.; Clare, D.K.; Winn, M.; Topf, M. Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. Methods 2016, 100, 42–49. [Google Scholar] [CrossRef]
- Pintilie, G.D.; Zhang, J.; Goddard, T.D.; Chiu, W.; Gossard, D.C. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010, 170, 427–438. [Google Scholar] [CrossRef]
- Park, Y.J.; Lacourse, K.D.; Cambillau, C.; DiMaio, F.; Mougous, J.D.; Veesler, D. Structure of the type VI secretion system TssK–TssF–TssG baseplate subcomplex revealed by cryo-electron microscopy. Nat. Commun. 2018, 9, 5385. [Google Scholar] [CrossRef] [Green Version]
- Bartesaghi, A.; Matthies, D.; Banerjee, S.; Merk, A.; Subramaniam, S. Structure of beta-galactosidase at 3.2-A resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 11709–11714. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human gamma-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Kamisetty, H.; Ovchinnikov, S.; Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. USA 2013, 110, 15674–15679. [Google Scholar] [CrossRef] [PubMed]
- Oteri, F.; Nadalin, F.; Champeimont, R.; Carbone, A. BIS2Analyzer: A server for co-evolution analysis of conserved protein families. Nucleic Acids Res. 2017, 45, W307–W314. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouvier, G.; Bardiaux, B.; Pellarin, R.; Rapisarda, C.; Nilges, M. Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution. Biomolecules 2022, 12, 1290. https://doi.org/10.3390/biom12091290
Bouvier G, Bardiaux B, Pellarin R, Rapisarda C, Nilges M. Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution. Biomolecules. 2022; 12(9):1290. https://doi.org/10.3390/biom12091290
Chicago/Turabian StyleBouvier, Guillaume, Benjamin Bardiaux, Riccardo Pellarin, Chiara Rapisarda, and Michael Nilges. 2022. "Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution" Biomolecules 12, no. 9: 1290. https://doi.org/10.3390/biom12091290
APA StyleBouvier, G., Bardiaux, B., Pellarin, R., Rapisarda, C., & Nilges, M. (2022). Building Protein Atomic Models from Cryo-EM Density Maps and Residue Co-Evolution. Biomolecules, 12(9), 1290. https://doi.org/10.3390/biom12091290