
Archaea as a Model System for Molecular Biology and Biotechnology

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Archaea as a Model System for Genetic Evolution
2.1. Genetic Tools
2.1.1. Transformation, Heterologous Protein Expression and Genetic Manipulation
2.1.2. Gene Knockout
2.2. Archaea as a Model System of Replication, Transcription, and Regulation of Gene Expression
2.2.1. DNA Replication and Repair
2.2.2. Transcription
2.2.3. Translational Recoding
3. Discovery and Biotechnological Applications of Archaeal Enzymes
3.1. Enzyme Discovery
3.2. Archaea Enzymes and Their Applications
3.2.1. Starch-Degrading Enzymes
3.2.2. Cellulose-Degrading Enzymes
3.2.3. Xylan-Degrading Enzymes
3.2.4. Chitin-Degrading Enzymes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, G.J.; Woese, C.R. Lessons from an Archaeal genome: What are we learning from Methanococcus jannaschii? Trends Genet. 1996, 12, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Eme, L.; Spang, A.; Lombard, J.; Stairs, C.W.; Ettema, T.J.G. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 2017, 15, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Straub, C.T.; Counts, J.A.; Nguyen, D.M.N.; Wu, C.H.; Zeldes, B.M.; Crosby, J.R.; Conway, J.M.; Otten, J.K.; Lipscomb, G.L.; Schut, G.J.; et al. Biotechnology of extremely thermophilic archaea. FEMS Microbiol. Rev. 2018, 42, 543–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quehenberger, J.; Shen, L.; Albers, S.V.; Siebers, B.; Spadiut, O. Sulfolobus—A Potential Key Organism in Future Biotechnology. Front. Microbiol. 2017, 8, 2474. [Google Scholar] [CrossRef] [Green Version]
- Curci, N.; Strazzulli, A.; Iacono, R.; De Lise, F.; Maurelli, L.; Di Fenza, M.; Cobucci-Ponzano, B.; Moracci, M. Xyloglucan Oligosaccharides Hydrolysis by Exo-Acting Glycoside Hydrolases from Hyperthermophilic Microorganism Saccharolobus solfataricus. Int. J. Mol. Sci. 2021, 22, 3325. [Google Scholar] [CrossRef]
- Iacono, R.; Cobucci-Ponzano, B.; De Lise, F.; Curci, N.; Maurelli, L.; Moracci, M.; Strazzulli, A. Spatial Metagenomics of Three Geothermal Sites in Pisciarelli Hot Spring Focusing on the Biochemical Resources of the Microbial Consortia. Molecules 2020, 25, 4023. [Google Scholar] [CrossRef]
- Strazzulli, A.; Cobucci-Ponzano, B.; Iacono, R.; Giglio, R.; Maurelli, L.; Curci, N.; Schiano-di-Cola, C.; Santangelo, A.; Contursi, P.; Lombard, V.; et al. Discovery of hyperstable carbohydrate-active enzymes through metagenomics of extreme environments. FEBS J. 2020, 287, 1116–1137. [Google Scholar] [CrossRef]
- Williams, T.A.; Foster, P.G.; Cox, C.J.; Embley, T.M. An archaeal origin of eukaryotes supports only two primary domains of life. Nature 2013, 504, 231–236. [Google Scholar] [CrossRef]
- Pérez-Arnaiz, P.; Dattani, A.; Smith, V.; Allers, T. Haloferax volcanii-a model archaeon for studying DNA replication and repair. Open Biol. 2020, 10, 200293. [Google Scholar] [CrossRef]
- Atomi, H.; Imanaka, T.; Fukui, T. Overview of the genetic tools in the Archaea. Front. Microbiol. 2012, 3, 337. [Google Scholar] [CrossRef] [Green Version]
- Zatopek, K.M.; Gardner, A.F.; Kelman, Z. Archaeal DNA replication and repair: New genetic, biophysical and molecular tools for discovering and characterizing enzymes, pathways and mechanisms. FEMS Microbiol. Rev. 2018, 42, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Berkner, S.; Ajon, M.; Driessen, A.J.; Lipps, G.; Albers, S.V. Expanding and understanding the genetic toolbox of the hyperthermophilic genus Sulfolobus. Biochem. Soc. Trans. 2009, 37, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.A.; Albers, S.V.; Atomi, H.; Allers, T. Model organisms for genetics in the domain Archaea: Methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 2011, 35, 577–608. [Google Scholar] [CrossRef] [Green Version]
- Lipscomb, G.L.; Stirrett, K.; Schut, G.J.; Yang, F.; Jenney, F.E.; Scott, R.A.; Adams, M.W.W.; Westpheling, J. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: Construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microb. 2011, 77, 2232–2238. [Google Scholar] [CrossRef] [Green Version]
- Grogan, D.W. Selectable mutant phenotypes of the extremely thermophilic archaebacterium Sulfolobus acidocaldarius. J. Bacteriol. 1991, 173, 7725–7727. [Google Scholar] [CrossRef] [Green Version]
- Martusewitsch, E.; Sensen, C.W.; Schleper, C. High spontaneous mutation rate in the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by transposable elements. J. Bacteriol. 2000, 182, 2574–2581. [Google Scholar] [CrossRef] [Green Version]
- Albers, S.V.; Driessen, A.J. Conditions for gene disruption by homologous recombination of exogenous DNA into the Sulfolobus solfataricus genome. Archaea 2008, 2, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.A.; Williams, S.A.; Santangelo, T.J. Thermococcus kodakarensis provides a versatile hyperthermophilic archaeal platform for protein expression. In Methods in Enzymology; Zvi Kelman, W., O’Dell, B., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 659, pp. 243–273. [Google Scholar]
- Li, Z.; Santangelo, T.J.; Cubonová, L.; Reeve, J.N.; Kelman, Z. Affinity purification of an archaeal DNA replication protein network. MBio 2010, 1, e00221-10. [Google Scholar] [CrossRef]
- Guss, A.M.; Rother, M.; Zhang, J.K.; Kulkarni, G.; Metcalf, W.W. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2008, 2, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Speed, M.C.; Burkhart, B.W.; Picking, J.W.; Santangelo, T.J. An Archaeal Fluoride-Responsive Riboswitch Provides an Inducible Expression System for Hyperthermophiles. Appl. Environ. Microbiol. 2018, 84, e02306-17. [Google Scholar] [CrossRef] [Green Version]
- Schocke, L.; Bräsen, C.; Siebers, B. Thermoacidophilic Sulfolobus species as source for extremozymes and as novel archaeal platform organisms. Curr. Opin. Biotechnol. 2019, 59, 71–77. [Google Scholar] [CrossRef]
- Atomi, H.; Reeve, J. Microbe profile: Thermococcus kodakarensis: The model hyperthermophilic archaeon. Microbiology 2019, 165, 1166–1168. [Google Scholar] [CrossRef]
- Burkhart, B.W.; Febvre, H.P.; Santangelo, T.J. Distinct physiological roles of the three ferredoxins encoded in the hyperthermophilic archaeon Thermococcus kodakarensis. MBio 2019, 10, e02807–e02818. [Google Scholar] [CrossRef] [Green Version]
- Takemasa, R.; Yokooji, Y.; Yamatsu, A.; Atomi, H.; Imanaka, T. Thermococcus kodakarensis as a host for gene expression and protein secretion. Appl. Environ. Microbiol. 2011, 77, 2392–2398. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, T.J.; Cubonová, L.U.; James, C.L.; Reeve, J.N. TFB1 or TFB2 is sufficient for Thermococcus kodakaraensis viability and for basal transcription in vitro. J. Mol. Biol. 2007, 367, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, T.J.; Reeve, J.N. Genetic tools and manipulations of the hyperthermophilic heterotrophic archaeon Thermococcus kodakarensis. In Extremophiles Handbook; Horikoshi, K., Ed.; Springer: Tokyo, Japan, 2010; pp. 567–582. [Google Scholar]
- Peng, N.; Han, W.; Li, Y.; Liang, Y.; She, Q. Genetic technologies for extremely thermophilic microorganisms of Sulfolobus, the only genetically tractable genus of crenarchaea. Sci. China Life Sci. 2017, 60, 370–385. [Google Scholar] [CrossRef]
- Matsubara, K.; Yokooji, Y.; Atomi, H.; Imanaka, T. Biochemical and genetic characterization of the three metabolic routes in Thermococcus kodakarensis linking glyceraldehyde 3-phosphate and 3-phosphoglycerate. Mol. Microbiol. 2011, 81, 1300–1312. [Google Scholar] [CrossRef]
- Yokooji, Y.; Tomita, H.; Atomi, H.; Imanaka, T. Pantoate kinase and phosphopantothenate synthetase, two novel enzymes necessary for CoA biosynthesis in the Archaea. J. Biol. Chem. 2009, 284, 28137–28145. [Google Scholar] [CrossRef]
- Borges, N.; Matsumi, R.; Imanaka, T.; Atomi, H.; Santos, H. Thermococcus kodakarensis mutants deficient in di-myo-inositol phosphate use aspartate to cope with heat stress. J. Bacteriol. 2010, 192, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Fukui, T.; Atomi, H.; Imanaka, T. Targeted gene disruption by ho- mologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 2003, 185, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Waege, I.; Schmid, G.; Thumann, S.; Thomm, M.; Hausner, W. Shuttle vector-based transformation system for Pyrococcus furiosus. Appl. Environ. Microbiol. 2010, 76, 3308–3313. [Google Scholar] [CrossRef] [Green Version]
- Schleper, C.; Kubo, K.; Zillig, W. The particle SSV1 from the extremely thermophilic archaeon Sulfolobus is a virus: Demonstration of infectivity and of transfection with viral DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7645–7649. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Kurosawa, N. Disruption of the gene encoding restriction endonuclease SuaI and development of a host-vector system for the thermoacidophilic archaeon Sulfolobus acidocaldarius. Extremophiles 2016, 20, 139–148. [Google Scholar] [CrossRef]
- Worthington, P.; Hoang, V.; Perez-Pomares, F.; Blum, P. Targeted disruption of the α-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 2003, 185, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Berkner, S.; Wlodkowski, A.; Albers, S.-V.; Lipps, G. Inducible and constitutive promoters for genetic systems in Sulfolobus acidocaldarius. Extremophiles 2010, 14, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Schelert, J.; Rudrappa, D.; Johnson, T.; Blum, P. Role of MerH in mercury resistance in the archaeon Sulfolobus solfataricus. Microbiology 2013, 159, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Szabó, Z.; Sani, M.; Groeneveld, M.; Zolghadr, B.; Schelert, J.; Albers, S.V.; Blum, P.; Boekema, E.J.; Driessen, A.J.M. Flagellar motility and structure in the hyperthermoacidophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 2007, 189, 4305–4309. [Google Scholar] [CrossRef] [Green Version]
- Zolghadr, B.; Weber, S.; Szabó, Z.; Driessen, A.J.M.; Albers, S.V. Identification of a system required for the functional surface localization of sugar binding proteins with class III signal peptides in Sulfolobus solfataricus. Mol. Microbiol. 2007, 64, 795–806. [Google Scholar] [CrossRef]
- Wong, J.H.Y.; Brown, J.A.; Suo, Z.; Blum, P.; Nohmi, T.; Ling, H. Structural insight into dynamic bypass of the major cisplatin-DNA adduct by Y-family polymerase Dpo4. EMBO J. 2010, 29, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Bitan-Banin, G.; Ortenberg, R.; Mevarech, M. Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 2003, 185, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Zhu, H.; Chen, Z.; Liang, Y.X.; She, Q. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 2009, 13, 735–746. [Google Scholar] [CrossRef]
- She, Q.; Zhang, C.; Deng, L.; Peng, N.; Chen, Z.; Liang, Y.X. Genetic analyses in the hyperthermophilic archaeon Sulfolobus islandicus. Biochem. Soc. Trans. 2009, 37, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Hong, Y.; Han, W.; Sheng, D.; Ni, J.; Hou, G.; Shen, Y. Single-stranded DNA binding activity of XPBI, but not XPBII, from Sulfolobus tokodaii causes double-stranded DNA melting. Extremophiles 2011, 15, 67–76. [Google Scholar] [CrossRef]
- Richards, J.D.; Cubeddu, L.; Roberts, J.; Liu, H.; White, M.F. The archaeal XPB protein is a ssDNA-dependent ATPase with a novel partner. J. Mol. Biol. 2008, 376, 634–644. [Google Scholar] [CrossRef]
- Albers, S.V.; Jarrell, K.F. The archaellum: How Archaea swim. Front. Microbiol. 2015, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Stachler, A.E.; Marchfelder, A. Gene Repression in Haloarchaea Using the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas I-B System. J. Biol. Chem. 2016, 291, 15226–15242. [Google Scholar] [CrossRef] [Green Version]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, I.A.; Wimmer, E.; Schleper, C. Heavily Armed Ancestors: CRISPR Immunity and Applications in Archaea with a Comparative Analysis of CRISPR Types in Sulfolobales. Biomolecules 2020, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, S.; Wang, Y.; Tian, X.; Zhao, P.; Xu, J.; Feng, M.; She, Q. CRISPR-Cas adaptive immune systems in Sulfolobales: Genetic studies and molecular mechanisms. Sci. China Life Sci. 2021, 64, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.; Shah, S.; Erdmann, S.; Liu, G.; Mousaei, M.; León-Sobrino, C.; Peng, W.; Gudbergsdottir, S.; Deng, L.; Vestergaard, G.; et al. CRISPR-Cas Adaptive Immune Systems of the Sulfolobales: Unravelling Their Complexity and Diversity. Life 2015, 5, 783–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebec, Z.; Manica, A.; Zhang, J.; White, M.F.; Schleper, C. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014, 42, 5280–5288. [Google Scholar] [CrossRef] [Green Version]
- Zink, I.A.; Fouqueau, T.; Tarrason Risa, G.; Werner, F.; Baum, B.; Bläsi, U.; Schleper, C. Comparative CRISPR type III-based knockdown of essential genes in hyperthermophilic Sulfolobales and the evasion of lethal gene silencing. RNA Biol. 2021, 18, 421–434. [Google Scholar] [CrossRef]
- Han, W.; Feng, X.; She, Q. Reverse gyrase functions in genome integrity maintenance by protecting DNA breaks in vivo. Int. J. Mol. Sci. 2017, 18, 1340. [Google Scholar] [CrossRef] [Green Version]
- Bassani, F.; Zink, I.A.; Pribasnig, T.; Wolfinger, M.T.; Romagnoli, A.; Resch, A.; Schleper, C.; Bläsi, U.; La Teana, A. Indications for a moonlighting function of translation factor aIF5A in the crenarchaeum Sulfolobus solfataricus. RNA Biol. 2019, 16, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liu, J.; Yang, H.; Xiang, H. DNA replication origins in archaea. Front. Microbiol. 2014, 5, 179. [Google Scholar] [CrossRef]
- Gehring, A.M.; Astling, D.P.; Matsumi, R.; Burkhart, B.W.; Kelman, Z.; Reeve, J.N.; Jones, K.L.; Santangelo, T.J. Genome Replication in Thermococcus kodakarensis Independent of Cdc6 and an Origin of Replication. Front. Microbiol. 2017, 8, 2084. [Google Scholar] [CrossRef]
- Le Breton, M.; Henneke, G.; Norais, C.; Flament, D.; Myllykallio, H.; Querellou, J.; Raffin, J.P. The heterodimeric primase from the euryarchaeon Pyrococcus abyssi: A multifunctional enzyme for initiation and repair? J. Mol. Biol. 2007, 374, 1172–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, H.; You, Z.; Arai, K. Control of DNA replication: Regulation and activation of eukaryotic replicative helicase, MCM. IUBMB Life 2005, 57, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Fujino, S.; Tomita, H.; Ogino, H.; Takao, K.; Daiyasu, H.; Kanai, T.; Atomi, H.; Ishino, Y. Biochemical and genetical analyses of the three mcm genes from the hyperthermophilic archaeon, Thermococcus kodakarensis. Genes Cells 2011, 16, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Liew, L.P.; Bell, S.D. The interplay of DNA binding, ATP hydrolysis and helicase activities of the archaeal MCM helicase. Biochem. J. 2011, 436, 409–414. [Google Scholar] [CrossRef]
- Kelman, L.M.; Kelman, Z. Archaeal DNA replication. Annu. Rev. Genet. 2014, 48, 71–97. [Google Scholar] [CrossRef]
- Cann, I.; Ishino, Y. Archaeal DNA replication: Identifying the pieces to solve a puzzle. Genetics 1999, 152, 1249–1267. [Google Scholar] [CrossRef]
- Grabowski, B.; Kelman, Z. Archaeal DNA replication: Eukaryal proteins in a bacterial context. Annu. Rev. Microbiol. 2003, 57, 487–516. [Google Scholar] [CrossRef]
- Kelman, Z.; White, M.F. Archaeal DNA replication and repair. Curr. Opin. Microbiol. 2005, 8, 669–676. [Google Scholar] [CrossRef]
- Walsh, E.; Eckert, K.A. Eukaryotic replicative DNA polymerases. In Nucleic Acid Polymerases; Murakami, K.S., Trakselis, M.A., Eds.; Nucleic Acids and Molecular Biology Series; 2014; Volume 30, pp. 17–41. [Google Scholar] [CrossRef]
- Čuboňová, L.; Richardson, T.; Burkhart, B.W.; Kelman, Z.; Connolly, B.A.; Reeve, J.N.; Santangelo, T.J. Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J. Bacteriol. 2013, 195, 2322–2328. [Google Scholar] [CrossRef]
- Li, Z.; Pan, M.; Santangelo, T.J.; Chemnitz, W.; Yuan, W.; Edwards, J.L.; Hurwitz, J.; Reeve, J.N.; Kelman, Z. A novel DNA nuclease is stimulated by association with the GINS complex. Nucleic Acids Res. 2011, 39, 6114–6123. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Guo, L.; Deng, L.; Wu, Y.; Liang, Y.; Huang, L.; She, Q. Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method. Microbiology 2010, 156 Pt 11, 3386–3397. [Google Scholar] [CrossRef] [PubMed]
- Woods, W.G.; Dyall-Smith, M.L. Construction and analysis of a recombination-deficient (radA) mutant of Haloferax volcanii. Mol. Microbiol. 1997, 23, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Fujikane, R.; Ishino, S.; Ishino, Y.; Forterre, P. Genetic analysis of DNA repair in the hyperthermophilic archaeon, Thermococcus kodakarensis. Genes Genet. Syst. 2010, 85, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; White, M.F. Hot and crispy: CRISPR-Cas systems in the hyperthermophile Sulfolobus solfataricus. Biochem. Soc. Trans. 2013, 41, 1422–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, W.; Chen, Y.; Guo, W.; Zhang, J.; Tang, H.; Xu, Z.; Zhang, H.; Tao, Y.; Wang, F.; et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016, 23, 1765–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, A.M.; Walker, J.E.; Santangelo, T.J. Transcription Regulation in Archaea. J. Bacteriol. 2016, 198, 1906–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, A.; Kanai, T.; Santangelo, T.J.; Tajiri, M.; Manabe, K.; Reeve, J.N.; Imanaka, T.; Murakami, K.S. Archaeal RNA polymerase subunits E and F are not required for transcription in vitro, but a Thermococcus kodakarensis mutant lacking subunit F is temperature-sensitive. Mol. Microbiol. 2008, 70, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Kanai, M.; Hanashiro, K.; Kim, S.H.; Hanai, S.; Boulares, A.H.; Miwa, M.; Fukasawa, K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 2007, 9, 1175–1183. [Google Scholar] [CrossRef]
- Hackley, R.K.; Schmid, A.K. Global Transcriptional Programs in Archaea Share Features with the Eukaryotic Environmental Stress Response. J. Mol. Biol. 2019, 431, 4147–4166. [Google Scholar] [CrossRef]
- Lemmens, L.; Maklad, H.R.; Bervoets, I.; Peeters, E. Transcription Regulators in Archaea: Homologies and Differences with Bacterial Regulators. J. Mol. Biol. 2019, 431, 4132–4146. [Google Scholar] [CrossRef]
- Villafane, A.; Voskoboynik, Y.; Ruhl, I.; Sannino, D.; Maezato, Y.; Blum, P.; Bini, E. CopR of Sulfolobus solfataricus represents a novel class of archaeal-specific copper-responsive activators of transcription. Microbiology 2011, 157, 2808–2817. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.K.; Pan, M.; Sharma, K.; Baliga, N.S. Two transcription factors are necessary for iron homeostasis in a salt-dwelling archaeon. Nucleic Acids Res. 2011, 39, 2519–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lie, T.J.; Leigh, J.A. A novel repressor of nif and glnA expression in the methanogenic archaeon Methanococcus maripaludis. Mol. Microbiol. 2003, 47, 235–246. [Google Scholar] [CrossRef] [PubMed]
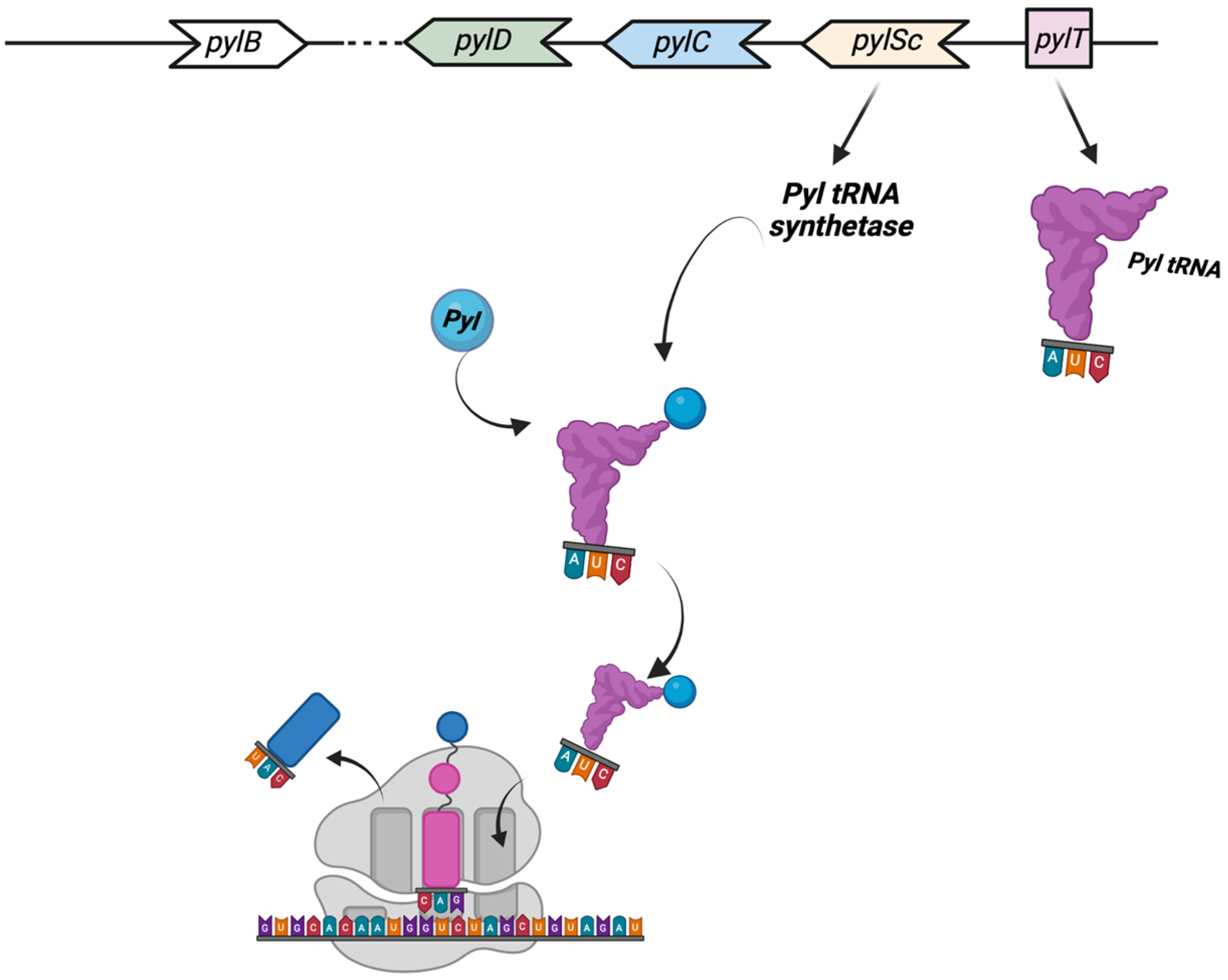
- Hao, B.; Gong, W.; Ferguson, T.K.; James, C.M.; Krzycki, J.A.; Chan, M.K. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 2002, 296, 1462–1466. [Google Scholar] [CrossRef]
- Srinivasan, G.; James, C.M.; Krzycki, J.A. Pyrrolysine encoded by UAG in Archaea: Charging of a UAG- decoding specialized tRNA. Science 2002, 296, 1459–1462. [Google Scholar] [CrossRef]
- Brugère, J.F.; Atkins, J.F.; O’Toole, P.W.; Borrel, G. Pyrrolysine in archaea: A 22nd amino acid encoded through a genetic code expansion. Emerg. Top. Life Sci. 2018, 2, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Gaston, M.A.; Zhang, L.; Green-Church, K.B.; Krzycki, J.A. The complete biosynthesis of the genetically encoded amino acid pyrrolysine fromlysine. Nature 2011, 471, 647–650. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, A.; Patel, A.; Soares, J.A.; Larue, R.C.; Zhang, J.K.; Metcalf, W.W.; Krzycki, J.A. Characterization of a Methanosarcina acetivorans mutant unable to translate UAG as pyrrolysine. Mol. Microbiol. 2006, 59, 56–66. [Google Scholar] [CrossRef]
- Lateef, O.M.; Akintubosun, M.O.; Olaoba, O.T.; Samson, S.O.; Adamczyk, M. Making Sense of “Nonsense” and More: Challenges and Opportunities in the Genetic Code Expansion, in the World of tRNA Modifications. Int. J. Mol. Sci. 2022, 23, 938. [Google Scholar] [CrossRef]
- Gesteland, R.F.; Atkins, J.F. Recoding: Dynamic reprogramming of translation. Annu. Rev. Biochem. 1996, 65, 741–768. [Google Scholar] [CrossRef]
- De Lise, F.; Strazzulli, A.; Iacono, R.; Curci, N.; Di Fenza, M.; Maurelli, L.; Moracci, M.; Cobucci-Ponzano, B. Programmed Deviations of Ribosomes From Standard Decoding in Archaea. Front. Microbiol. 2021, 12, 688061. [Google Scholar] [CrossRef] [PubMed]
- Baranov, P.V.; Gesteland, R.F.; Atkins, J.F. Recoding: Translational bifurcations in gene expression. Gene 2002, 286, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Cobucci-Ponzano, B.; Trincone, A.; Giordano, A.; Rossi, M.; Moracci, M. Identification of an archaeal α-L-fucosidase encoded by an interrupted gene. Production of a functional enzyme by mutations mimicking programmed -1 frameshifting. J. Biol. Chem. 2003, 278, 14622–14631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobucci-Ponzano, B.; Rossi, M.; Moracci, M. Recoding in archaea. Mol. Microbiol. 2005, 55, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Cobucci-Ponzano, B.; Conte, F.; Benelli, D.; Londei, P.; Flagiello, A.; Monti, M.; Pucci, P.; Rossi, M.; Moracci, M. The gene of an archaeal alpha-L-fucosidase is expressed by translational frameshifting. Nucleic Acids Res. 2006, 34, 4258–4268. [Google Scholar] [CrossRef] [Green Version]
- De Lise, F.; Iacono, R.; Strazzulli, A.; Giglio, R.; Curci, N.; Maurelli, L.; Avino, R.; Carandente, A.; Caliro, S.; Tortora, A.; et al. Transcript Regulation of the Recoded Archaeal α-L-Fucosidase In Vivo. Molecules 2021, 26, 1861. [Google Scholar] [CrossRef]
- Rother, M.; Krzycki, J.A. Selenocysteine, pyrrolysine, and the unique energy metabolism of methanogenic archaea. Archaea 2010, 2010, 453642. [Google Scholar] [CrossRef] [Green Version]
- Rother, M.; Quitzke, V. Selenoprotein synthesis and regulation in Archaea. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2451–2462. [Google Scholar] [CrossRef]
- Rother, M.; Mathes, I.; Lottspeich, F.; Böck, A. Inactivation of the selB gene in Methanococcus maripaludis: Effect on synthesis of selenoproteins and their sulfur-containing homologs. J. Bacteriol. 2003, 185, 107–114. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Korniy, N.; Klimova, M.; Karki, P.; Peng, B.-Z.; Senyushkina, T.; Belardinelli, R.; Maracci, C.; Wohlgemuth, I.; Samatova, E.; et al. Translational recoding: Canonical translation mechanisms reinterpreted. Nucleic Acids Res. 2020, 48, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Cobucci-Ponzano, B.; Rossi, M.; Moracci, M. Translational recoding in archaea. Extremophiles 2012, 16, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Cobucci-Ponzano, B.; Trincone, A.; Giordano, A.; Rossi, M.; Moracci, M. Identification of the catalytic nucleophile of the family 29 α-L-fucosidase from Sulfolobus solfataricus via chemical rescue of an inactive mutant. Biochemistry 2003, 42, 9525–9531. [Google Scholar] [CrossRef] [PubMed]
- Cobucci-Ponzano, B.; Mazzone, M.; Rossi, M.; Moracci, M. Probing the catalytically essential residues of the α-L-fucosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. Biochemistry 2005, 44, 6331–6342. [Google Scholar] [CrossRef] [PubMed]
- Rosano, C.; Zuccotti, S.; Cobucci-Ponzano, B.; Mazzone, M.; Rossi, M.; Moracci, M.; Petoukhov, M.V.; Svergun, D.I.; Bolognesi, M. Structural characterization of the nonameric assembly of an Archaeal alpha-L-fucosidase by synchrotron small angle X-ray scattering. Biochem. Biophys. Res. Commun. 2004, 320, 176–182. [Google Scholar] [CrossRef]
- Raddadi, N.; Cherif, A.; Daffonchio, D.; Mohamed, N.; Fava, F. Biotechnological applications of extremophiles, extremozymes and extremolytes. Appl. Microbiol. Biotechnol. 2015, 99, 7907–7913. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, A.; Arun, K.B.; Binod, P.; Sirohi, R.; Tarafdar, A.; Reshmy, R.; Awasthi, M.K.; Sindhu, R. Design of Novel Enzyme Biocatalysts for Industrial Bioprocess: Harnessing the Power of Protein Engineering, High Throughput Screening and Synthetic Biology. Bioresour. Technol. 2021, 325, 124617. [Google Scholar] [CrossRef]
- Research, B. BCC Research Report. In Global Markets for Enymes in Industrial Applications; Bio030L; BCC Research LLC: Boston, MA, USA, 2021. [Google Scholar]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system-From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef]
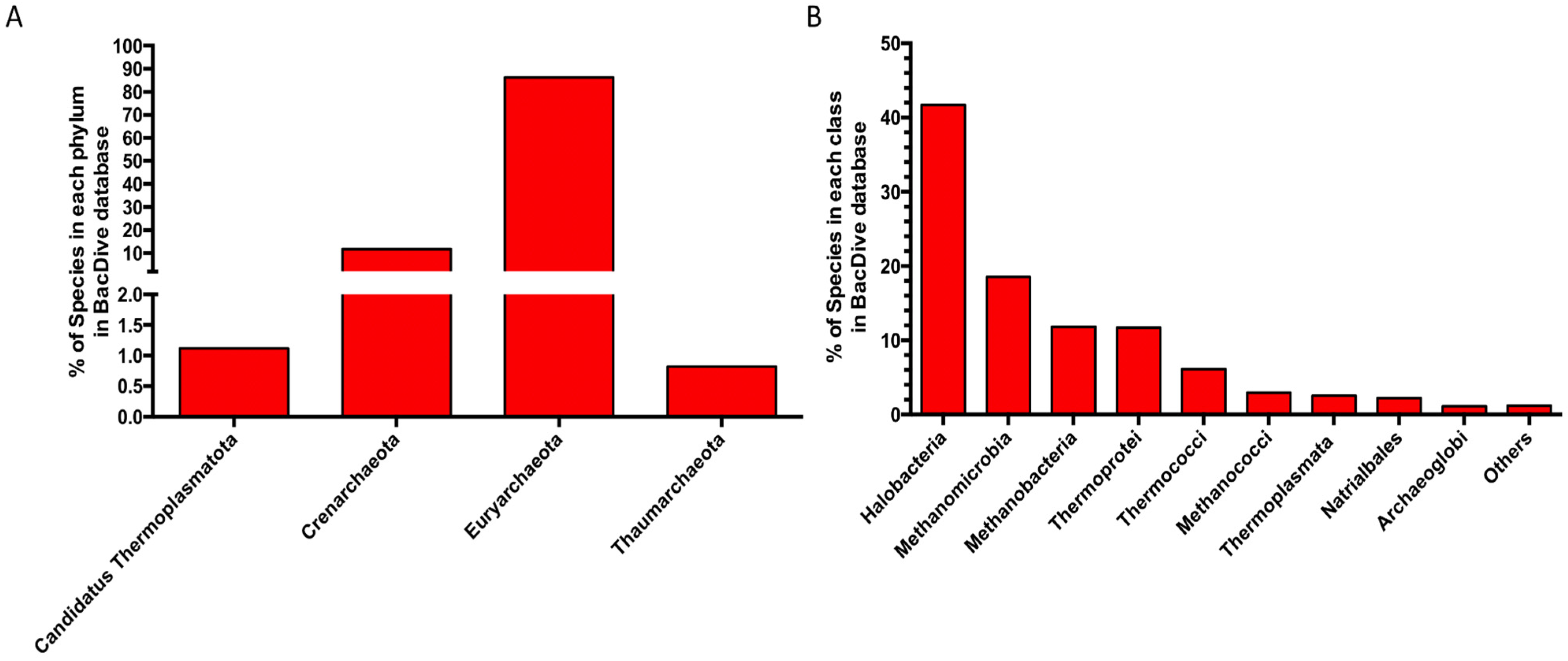
- Reimer, L.C.; Vetcininova, A.; Carbasse, J.S.; Sohngen, C.; Gleim, D.; Ebeling, C.; Overmann, J. BacDive in 2019: Bacterial phenotypic data for High-throughput biodiversity analysis. Nucleic Acids Res. 2019, 47, D631–D636. [Google Scholar] [CrossRef] [Green Version]
- Cross, K.L.; Campbell, J.H.; Balachandran, M.; Campbell, A.G.; Cooper, C.J.; Griffen, A.; Heaton, M.; Joshi, S.; Klingeman, D.; Leys, E.; et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol. 2019, 37, 1314–1321. [Google Scholar] [CrossRef]
- Uchiyama, T.; Miyazaki, K. Functional metagenomics for enzyme discovery: Challenges to efficient screening. Curr. Opin. Biotechnol. 2009, 20, 616–622. [Google Scholar] [CrossRef]
- Zaparucha, A.; de Berardinis, V.; Vaxelaire-Vergne, C. Genome Mining for Enzyme Discovery. In Modern Biocatalysis; Catalysis Series; RSC Publishing: London, UK, 2018; Chapter 1; pp. 1–27. [Google Scholar]
- Cobucci-Ponzano, B.; Aurilia, V.; Riccio, G.; Henrissat, B.; Coutinho, P.M.; Strazzulli, A.; Padula, A.; Corsaro, M.M.; Pieretti, G.; Pocsfalvi, G.; et al. A new archaeal beta-glycosidase from Sulfolobus solfataricus: Seeding a novel retaining beta-glycan-specific glycoside hydrolase family along with the human non-lysosomal glucosylceramidase GBA2. J. Biol. Chem. 2010, 285, 20691–20703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, M.C.; Cobucci-Ponzano, B.; Carpentieri, A.; Henrissat, B.; Rossi, M.; Amoresano, A.; Moracci, M. The identification and molecular characterization of the first archaeal bifunctional exo-β-glucosidase/N-acetyl-β-glucosaminidase demonstrate that family GH116 is made of three functionally distinct subfamilies. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Iacono, R.; Strazzulli, A.; Maurelli, L.; Curci, N.; Casillo, A.; Corsaro, M.M.; Moracci, M.; Cobucci-Ponzano, B. GlcNAc De-N-Acetylase from the Hyperthermophilic Archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 2019, 85, e01879-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurelli, L.; Giovane, A.; Esposito, A.; Moracci, M.; Fiume, I.; Rossi, M.; Morana, A. Evidence that the xylanase activity from Sulfolobus solfataricus Oalpha is encoded by the endoglucanase precursor gene (sso1354) and characterization of the associated cellulase activity. Extremophiles 2008, 12, 689–700. [Google Scholar] [CrossRef]
- Niehaus, F.; Peters, A.; Groudieva, T.; Antranikian, G. Cloning, expression and biochemical characterisation of a unique thermostable pullulan-hydrolysing enzyme from the hyperthermophilic archaeon Thermococcus aggregans. FEMS Microbiol. Lett. 2000, 190, 223–229. [Google Scholar] [CrossRef]
- De Castro, M.E.; Rodriguez-Belmonte, E.; Gonzalez-Siso, M.I. Metagenomics of Thermophiles with a Focus on Discovery of Novel Thermozymes. Front. Microbiol. 2016, 7, 1521. [Google Scholar] [CrossRef] [Green Version]
- Strazzulli, A.; Fusco, S.; Cobucci-Ponzano, B.; Moracci, M.; Contursi, P. Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev. Env. Sci. Bio./Technol. 2017, 16, 425–454. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.; Jain, K.; Madamwar, D. Functional Metagenomics. In Current Developments in Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2017; pp. 27–43. [Google Scholar]
- Iacono, R.; Strazzulli, A.; Giglio, R.; Bitetti, F.; Cobucci-Ponzano, B.; Moracci, M. Valorization of Biomasses from Energy Crops for the Discovery of Novel Thermophilic Glycoside Hydrolases through Metagenomic Analysis. Int. J. Mol. Sci. 2022, 23, 10505. [Google Scholar] [CrossRef]
- Liew, K.J.; Liang, C.H.; Lau, Y.T.; Yaakop, A.S.; Chan, K.G.; Shahar, S.; Shamsir, M.S.; Goh, K.M. Thermophiles and carbohydrate-active enzymes (CAZymes) in biofilm microbial consortia that decompose lignocellulosic plant litters at high temperatures. Sci. Rep. 2022, 12, 2850. [Google Scholar] [CrossRef]
- Mirete, S.; Morgante, V.; Gonzalez-Pastor, J.E. Functional metagenomics of extreme environments. Curr. Opin. Biotechnol. 2016, 38, 143–149. [Google Scholar] [CrossRef]
- Zatopek, K.M.; Fossa, S.L.; Bilotti, K.; Caffrey, P.J.; Chuzel, L.; Gehring, A.M.; Lohman, G.J.S.; Taron, C.H.; Gardner, A.F. Capillary Electrophoresis-Based Functional Genomics Screening to Discover Novel Archaeal DNA Modifying Enzymes. Appl. Environ. Microbiol. 2022, 88, e0213721. [Google Scholar] [CrossRef] [PubMed]
- Chuzel, L.; Ganatra, M.B.; Rapp, E.; Henrissat, B.; Taron, C.H. Functional metagenomics identifies an exosialidase with an inverting catalytic mechanism that defines a new glycoside hydrolase family (GH156). J. Biol. Chem. 2018, 293, 18138–18150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, A.; Zhou, J.; Pham, V.T.T.; Haugen, T.; Zeiny, M.E.; Aarstad, O.; Liebl, W.; Wentzel, A.; Liles, M.R. Novel archaeal thermostable cellulases from an oil reservoir metagenome. AMB Express 2017, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leis, B.; Heinze, S.; Angelov, A.; Pham, V.T.; Thurmer, A.; Jebbar, M.; Golyshin, P.N.; Streit, W.R.; Daniel, R.; Liebl, W. Functional Screening of Hydrolytic Activities Reveals an Extremely Thermostable Cellulase from a Deep-Sea Archaeon. Front. Bioeng. Biotechnol. 2015, 3, 95. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.N.; Cheng, J.; Engel, K.; Neufeld, J.D.; Charles, T.C. Current and future resources for functional metagenomics. Front. Microbiol. 2015, 6, 1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champdore, M.D.; Staiano, M.; Rossi, M.; D’Auria, S. Proteins from extremophiles as stable tools for advanced biotechnolog- ical applications of high social interest. J. R. Soc. Interface 2007, 4, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, S.B. Rare codon clusters at 5′-end influence heterologous expression of archaeal gene in Escherichia coli. Protein Expr. Purif. 2006, 50, 49–57. [Google Scholar] [CrossRef]
- Dadwal, A.; Sharma, S.; Satyanarayana, T. Thermostable cellulose saccharifying microbial enzymes: Characteristics, recent advances and biotechnological applications. Int. J. Biol. Macromol. 2021, 188, 226–244. [Google Scholar] [CrossRef]
- Suleiman, M.; Krüger, A.; Antranikian, G. Biomass-degrading glycoside hydrolases of archaeal origin. Biotechnol. Biofuels 2020, 13, 153. [Google Scholar] [CrossRef]
- Van den Burg, B. Extremophiles as a source for novel enzymes. Curr. Opin. Microbiol. 2003, 6, 213–218. [Google Scholar] [CrossRef]
- Amin, K.; Tranchimand, S.; Benvegnu, T.; Abdel-Razzak, Z.; Chamieh, H. Glycoside Hydrolases and Glycosyltransferases from Hyperthermophilic Archaea: Insights on Their Characteristics and Applications in Biotechnology. Biomolecules 2021, 11, 1557. [Google Scholar] [CrossRef] [PubMed]
- Van der Maarel, M.J.; van der Veen, B.; Uitdehaag, J.C.; Leemhuis, H.; Dijkhuizen, L. Properties and applications of starch-converting enzymes of the alpha-amylase family. J. Biotechnol. 2002, 94, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.; Nigam, P.; Soccol, C.R.; Soccol, V.T.; Singh, D.; Mohan, R. Advances in microbial amylases. Biotechnol. Appl. Biochem. 2000, 31, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Cabrera, M.Á.; Blamey, J.M. Biotechnological applications of archaeal enzymes from extreme environments. Biol. Res. 2018, 51, 37. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Govil, T.; Saxena, P.; Thakur, P.; Narayanan, A.; Sani, R.K. Extremozymes and Their Applications; Academic Press: Cambridge, MA, USA, 2022; Chapter 1; pp. 1–39. ISBN 9780323902748. [Google Scholar] [CrossRef]
- Chang, S.T.; Parker, K.N.; Bauer, M.W.; Kelly, R.M. alpha-Glucosidase from Pyrococcus furiosus. Methods Enzymol. 2001, 330, 260–269. [Google Scholar] [CrossRef]
- Galichet, A.; Belarbi, A. Cloning of an α-glucosidase gene from Thermococcus hydrothermalis by functional complementation of a Saccharomyces cerevisiae mal11 mutant strain. FEBS Lett. 1999, 458, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, M.; Golyshina, O.V.; Plou, F.J.; Timmis, K.N.; Golyshin, P.N. A novel alpha-glucosidase from the acidophilic archaeon Ferroplasma acidiphilum strain Y with high transglycosylation activity and an unusual catalytic nucleophile. Biochem. J. 2005, 391, 269–276. [Google Scholar] [CrossRef]
- Angelov, A.; Putyrski, M.; Liebl, W. Molecular and biochemical characterization of alpha-glucosidase and alpha-mannosidase and their clustered genes from the thermoacidophilic archaeon Picrophilus torridus. J. Bacteriol. 2006, 188, 7123–7131. [Google Scholar] [CrossRef] [Green Version]
- Akintunde, M.O.; Adebayo-Tayo, B.C.; Ishola, M.M.; Zamani, A.; Horváth, I.S. Bacterial Cellulose Production from agricultural Residues by two Komagataeibacter sp. Strains. Bioengineered 2022, 13, 10010–10025. [Google Scholar] [CrossRef]
- Prasad, R.K.; Chatterjee, S.; Sharma, S.; Mazumder, P.B.; Vairale, M.G.; Raju, P.S. Insect Gut Bacteria and Their Potential Application in Degradation of Lignocellulosic Biomass: A Review. In Bioremediation: Applications for Environmental Protection and Management; Varjani, S.J., Agarwal, A.K., Gnansounou, E., Gurunathan, B., Eds.; Springer: Singapore, 2018; pp. 277–299. [Google Scholar] [CrossRef]
- Graham, J.; Clark, M.; Nadler, D.; Huffer, S.; Chokhawala, H.A.; Rowland, S.E.; Blanch, H.W.; Clark, D.S.; Robb, F.T. Identification and characterization of a multidomain hyperthermophilic cellulase from an archaeal enrichment. Nat. Commun. 2011, 2, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, A.; Walsh, G. Expression and characterisation of a thermophilic endo-1,4-β-glucanase from Sulfolobus shibatae of potential industrial application. Mol. Biol. Rep. 2018, 45, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Lecourt, M.; Meyer, V.; Sigoillot, J.C.; Petit-Conil, M. Energy reduction of refining by cellulases. Holzforschung 2010, 64, 4416. [Google Scholar] [CrossRef]
- Niyonzima, F.N.; More, S.S. Purification and characterization of detergent-compatible protease from Aspergillus terreus gr. 3 Biotech 2015, 5, 6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.N.; Luna, I.Z.; Islam, M.M.; Sharmeen, S.; Salem, K.S.; Rashid, T.U.; Zaman, A.; Haque, P.; Rahman, M.M. Cellulase in waste management applications. In New and Future Developments in Microbial Biotechnology and Bioengineering; Microbial Cellulase System Properties and Applications; Elsevier: Amsterdam, The Netherlands, 2016; pp. 237–256. [Google Scholar]
- Singhania, R.R.; Sukumaran, R.K.; Patel, A.K.; Larroche, C.; Pandey, A. Advancement and comparative profiles in the production technologies using solid- state and submerged fermentation for microbial cellulases. Enzym. Microb. Technol. 2010, 46, 541–549. [Google Scholar] [CrossRef]
- Patel, A.K.; Singhania, R.R.; Sim, S.J.; Pandey, A. Thermostable cellulases: Current status and perspectives. Bioresour. Technol. 2019, 279, 385–392. [Google Scholar] [CrossRef]
- Ninawe, S.; Kapoor, M.; Kuhad, R.C. Purification and characterization of extracellular xylanase from Streptomyces cyaneus SN32. Bioresour. Technol. 2008, 99, 1252–1258. [Google Scholar] [CrossRef]
- Kumar, V.; Verma, D.; Satyanarayana, T. Extremophilic bacterial xylanases: Production, characteristics and applications. Curr. Biotechnol. 2013, 2, 380–399. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Kumar, B.; Verma, P. A detailed overview of xylanases: An emerging biomolecule for current and future prospective. Bioresour. Bioprocess. 2019, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Cobucci-Ponzano, B.; Strazzulli, A.; Iacono, R.; Masturzo, G.; Giglio, R.; Rossi, M.; Moracci, M. Novel thermophilic hemicellulases for the conversion of lignocellulose for second generation biorefineries. Enzym. Microb. Technol. 2015, 78, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Satyanarayana, T. Production of cellulase-free xylanase by the recombinant Bacillus subtilis and its applicability in paper pulp bleaching. Biotechnol. Prog. 2013, 29, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Passarinho, A.T.P.; Ventorim, R.Z.; Maitan-Alfenas, G.P.; de Oliveira, E.B.; Guimaraes, V.M. Engineered GH11 xylanases from Orpinomyces sp. PC-2 improve techno-functional properties of bread dough. J. Sci. Food Agric. 2019, 99, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B. Classification of chitinases modules. EXS 1999, 87, 137. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.; Gupta, R.D. Chitinases from bacteria to human: Properties, applications, and future perspectives. Enzym. Res. 2015, 2015, 791907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, T.; Ishikawa, K. Analysis of the hyperthermophilic chitinase from Pyrococcus furiosus: Activity toward crystalline chitin. Biosci. Biotechnol. Biochem. 2006, 70, 1696–1701. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Fukui, T.; Imanaka, T. Different cleavage specificities of the dual catalytic domains in chitinase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Biol. Chem. 2001, 276, 35629–35635. [Google Scholar] [CrossRef] [Green Version]
- Andronopoulou, E.; Vorgias, C.E. Purification and characterization of a new hyperthermostable, allosamidin-insensitive and denaturation-resistant chitinase from the hyperthermophilic archaeon Thermococcus chitonophagus. Extremophiles 2003, 7, 4353. [Google Scholar] [CrossRef]
- Le, B.; Yang, S.H. Microbial chitinases: Properties, current state and biotechnological applications. World J. Microbiol. Biotechnol. 2019, 35, 144. [Google Scholar] [CrossRef]
- Rameshthangam, P.; Solairaj, D.; Arunachalam, G.; Ramasamy, P. Chitin and chitinases: Biomedical and environmental applications of chitin and its derivatives. J. Enzym. 2018, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Annamalai, N.; Rajeswari, M.V.; Vijayalakshmi, S.; Balasubramanian, T. Purification and characterization of chitinase from Alcaligenes faecalis AU02 by utilizing marine wastes and its antioxidant activity. Ann. Microbiol. 2011, 61, 8017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, G.M.; Sukumaran, R.K.; Sindhu, R.; Binod, P.; Pandey, A. Green remediation of the potential hazardous shellfish wastes generated from the processing industries and their bioprospecting. Environ. Technol. Innov. 2021, 24, 101979. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Subclass | Description | Entries in BRENDA Database | % of BRENDA Archaeal Entries |
|---|---|---|---|
| 1.1.1.- | Oxidoreductases acting on the CH-OH group of donors with NAD+ or NADP+ as acceptor | 259 | 6.58 |
| 3.2.1.- | Glycosidases | 206 | 5.23 |
| 2.7.7.- | Nucleotidyltransferases | 167 | 4.24 |
| 2.1.1.- | Methyltransferases | 151 | 3.83 |
| 2.7.1.- | Phosphotransferases with an alcohol group as acceptor | 151 | 3.83 |
| 6.1.1.- | Ligases forming aminoacyl-tRNA and related compounds | 112 | 2.84 |
| 2.5.1.- | Transferases active on alkyl or aryl groups, other than methyl | 98 | 2.49 |
| 4.2.1.- | Hydro-lyases | 97 | 2.46 |
| 1.2.7.- | Oxidoreductases with an iron-sulfur protein as acceptor | 78 | 1.98 |
| 4.1.1.- | Carboxy-lyases | 74 | 1.88 |
| 2.4.99.- | Glycosyltransferases transferring other glycosyl groups | 72 | 1.83 |
| 3.1.1.- | Carboxylic-ester hydrolases | 68 | 1.73 |
| 2.4.1.- | Hexosyltransferases | 64 | 1.63 |
| 3.6.4.- | Hydrolases acting on acid anhydrides to facilitate cellular and subcellular movement | 63 | 1.60 |
| 1.2.1.- | Oxidoreductases acting on the aldehyde or oxo group of donors with NAD+ or NADP+ as acceptor | 60 | 1.52 |
| 2.3.1.- | Acyltransferases transferring groups other than aminoacyl groups | 57 | 1.45 |
| 3.1.3.- | Phosphoric-monoester hydrolases | 54 | 1.37 |
| 6.2.1.- | Acid-thiol ligases | 53 | 1.35 |
| 2.6.1.- | Transaminases | 52 | 1.32 |
| 1.4.1.- | Oxidoreductases Acting on the CH-NH2 group of donors with NAD+ or NADP+ as acceptor | 51 | 1.30 |
| 2.4.2.- | Pentosyltransferases | 51 | 1.30 |
| 4.1.2.- | Aldehyde-lyases | 51 | 1.30 |
| 6.5.1.- | Ligases that form phosphoric-ester bonds | 51 | 1.30 |
| 5.6.2.- | Enzymes altering nucleic acid conformation | 49 | 1.24 |
| 3.5.4.- | Hydrolases | 48 | 1.22 |
| 3.5.1.- | Acting on carbon-nitrogen bonds, other than peptide bonds in cyclic amidines | 46 | 1.17 |
| 6.3.4.- | Ligases Forming carbon-nitrogen bonds other carbon-nitrogen ligases | 45 | 1.14 |
| 3.6.1.- | Hydrolases acting on acid anhydrides in phosphorus-containing anhydrides | 44 | 1.12 |
| 5.3.1.- | Intramolecular oxidoreductases Interconverting aldoses and ketoses, and related compounds | 44 | 1.12 |
| 2.7.4.- | Phosphotransferases with a phosphate group as acceptor | 43 | 1.09 |
| 3.4.21.- | Serine endopeptidases | 42 | 1.07 |
| 2.8.4.- | Transferases transferring alkylthio groups | 41 | 1.04 |
| 3.1.26.- | Endoribonucleases producing 5′-phosphomonoesters | 41 | 1.04 |
| 6.3.2.- | Peptide synthases | 40 | 1.02 |
| Others | Sub-sub classes with relative abundance < 1% | 1315 | 33.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lise, F.; Iacono, R.; Moracci, M.; Strazzulli, A.; Cobucci-Ponzano, B. Archaea as a Model System for Molecular Biology and Biotechnology. Biomolecules 2023, 13, 114. https://doi.org/10.3390/biom13010114
De Lise F, Iacono R, Moracci M, Strazzulli A, Cobucci-Ponzano B. Archaea as a Model System for Molecular Biology and Biotechnology. Biomolecules. 2023; 13(1):114. https://doi.org/10.3390/biom13010114
Chicago/Turabian StyleDe Lise, Federica, Roberta Iacono, Marco Moracci, Andrea Strazzulli, and Beatrice Cobucci-Ponzano. 2023. "Archaea as a Model System for Molecular Biology and Biotechnology" Biomolecules 13, no. 1: 114. https://doi.org/10.3390/biom13010114
APA StyleDe Lise, F., Iacono, R., Moracci, M., Strazzulli, A., & Cobucci-Ponzano, B. (2023). Archaea as a Model System for Molecular Biology and Biotechnology. Biomolecules, 13(1), 114. https://doi.org/10.3390/biom13010114