
Laboratory and Metabolomic Fingerprint in Heart Failure with Preserved Ejection Fraction: From Clinical Classification to Biomarker Signature


Abstract
:1. Introduction
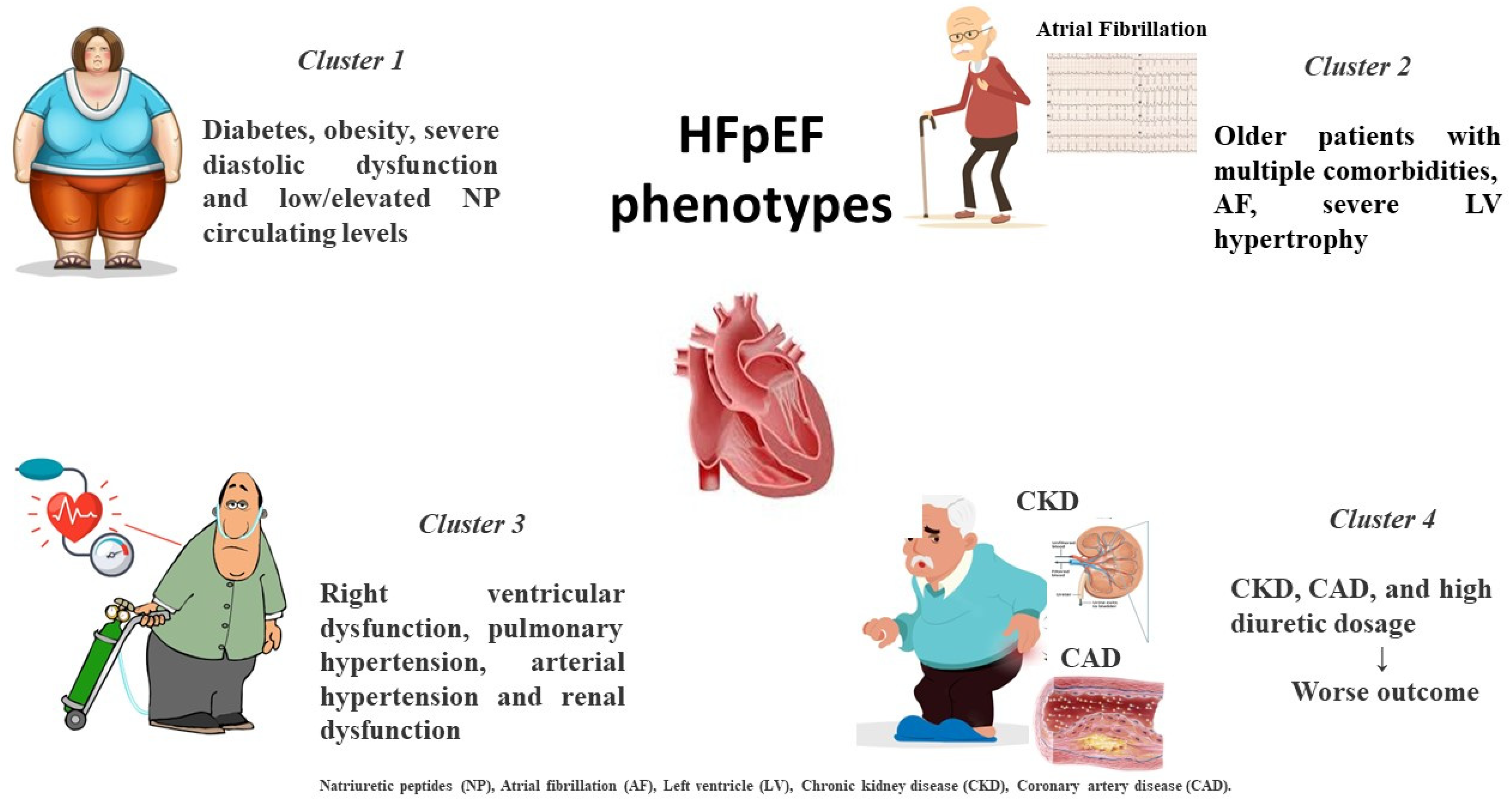
2. Different HFpEF Phenogroups
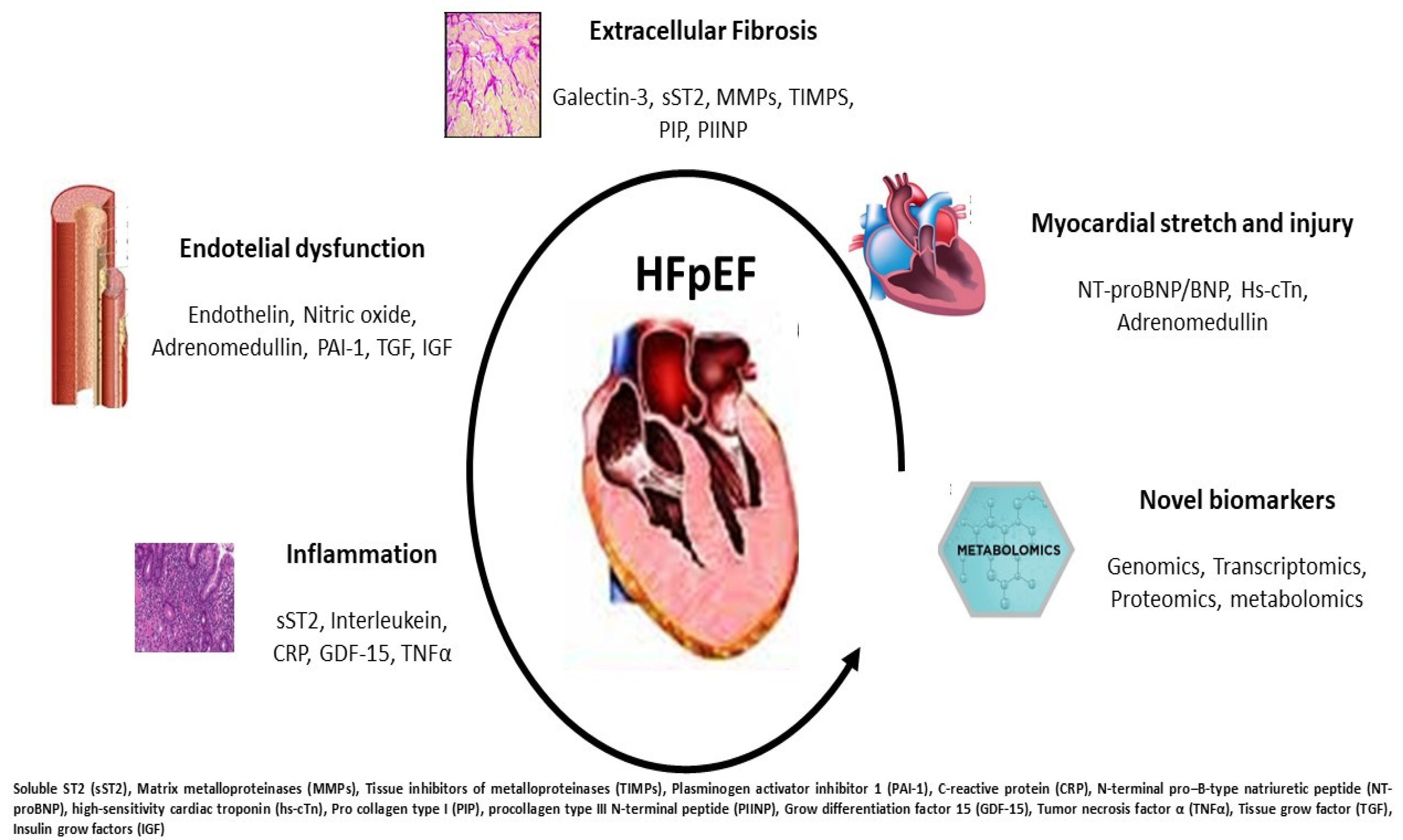
3. Current Biomarkers in HFpEF
4. Metabolomic Signature
5. Circulating MicroRNA Evidence
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lewis, G.A.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; Ahmed, F.; McDonagh, T.A.; Miller, C.A. Biological Phenotypes of Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Paulus, W.J. Heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and treatment. Eur. Heart J. 2011, 32, 670–679. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Xu, Y.; Yue, F.; Zhang, C.; Chen, C. Quality of inclusion criteria in the registered clinical trials of heart failure with preserved ejection fraction: Is it time for a change? Int. J. Cardiol. 2018, 254, 210–214. [Google Scholar] [CrossRef]
- Shah, A.M.; Shah, S.J.; Anand, I.S.; Sweitzer, N.K.; O’Meara, E.; Heitner, J.F.; Sopko, G.; Li, G.; Assmann, S.F.; McKinlay, S.M.; et al. Cardiac structure and function in heart failure with preserved ejection fraction: Baseline findings from the echocardiographic study of the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist trial. Circ. Heart Fail. 2014, 7, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.M.; Cikes, M.; Prasad, N.; Li, G.; Getchevski, S.; Claggett, B.; Rizkala, A.; Lukashevich, I.; O’Meara, E.; Ryan, J.J.; et al. Echocardiographic Features of Patients with Heart Failure and Preserved Left Ventricular Ejection Fraction. J. Am. Coll. Cardiol. 2019, 74, 2858–2873. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Katz, D.H.; Deo, R.C. Phenotypic spectrum of heart failure with preserved ejection fraction. Heart Fail. Clin. 2014, 10, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.P.; Mentz, R.J.; Mebazaa, A.; Voors, A.A.; Butler, J.; Roessig, L.; Fiuzat, M.; Zannad, F.; Pitt, B.; O’Connor, C.M.; et al. Patient selection in heart failure with preserved ejection fraction clinical trials. J. Am. Coll. Cardiol. 2015, 65, 1668–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaduganathan, M.; Michel, A.; Hall, K.; Mulligan, C.; Nodari, S.; Shah, S.J.; Senni, M.; Triggiani, M.; Butler, J.; Gheorghiade, M. Spectrum of epidemiological and clinical findings in patients with heart failure with preserved ejection fraction stratified by study design: A systematic review. Eur. J. Heart Fail. 2016, 18, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J. Innovative Clinical Trial Designs for Precision Medicine in Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Transl. Res. 2017, 10, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Palazzuoli, A.; Caravita, S.; Paolillo, S.; Ghio, S.; Tocchetti, C.G.; Ruocco, G.; Correale, M.; Ambrosio, G.; Perrone Filardi, P.; Senni, M.; et al. Current gaps in HFpEF trials: Time to reconsider patients’ selection and to target phenotypes. Prog. Cardiovasc. Dis. 2021, 67, 89–97. [Google Scholar] [CrossRef]
- Shah, A.M.; Claggett, B.; Sweitzer, N.K.; Shah, S.J.; Anand, I.S.; O’Meara, E.; Desai, A.S.; Heitner, J.F.; Li, G.; Fang, J.; et al. Cardiac structure and function and prognosis in heart failure with preserved ejection fraction: Findings from the echocardiographic study of the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) Trial. Circ. Heart Fail. 2014, 7, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Gottdiener, J.S.; Hetzel, S.J.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Baicu, C.F.; Massie, B.M.; Carson, P.E.; I-PRESERVE Investigator. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation 2011, 124, 2491–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, R.T.; Jhund, P.S.; Castagno, D.; Hawkins, N.M.; Petrie, M.C.; McMurray, J.J. What have we learned about patients with heart failure and preserved ejection fraction from DIG-PEF, CHARM-preserved, and I-PRESERVE? J. Am. Coll. Cardiol. 2012, 60, 2349–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, A.; Senni, M.; Barbati, G.; Greene, S.J.; Poli, S.; Zambon, E.; Di Nora, C.; Cioffi, G.; Tarantini, L.; Gavazzi, A.; et al. Prevalence and prognostic impact of non-cardiac co-morbidities in heart failure outpatients with preserved and reduced ejection fraction: A community-based study. Eur. J. Heart Fail. 2018, 20, 1257–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.B.; Schrauben, S.J.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Yarde, M.; Wang, Z.; Bhattacharya, P.T.; Chirinos, D.A.; et al. Clinical Phenogroups in Heart Failure with Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.C.; Deo, R.C. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Uijl, A.; Savarese, G.; Vaartjes, I.; Dahlström, U.; Brugts, J.J.; Linssen, G.C.M.; van Empel, V.; Brunner-La Rocca, H.P.; Asselbergs, F.W.; Lund, L.H.; et al. Identification of distinct phenotypic clusters in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 973–982. [Google Scholar] [CrossRef]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; et al. Identifying Pathophysiological Mechanisms in Heart Failure with Reduced versus Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2018, 72, 1081–1090. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Beltrami, M. Are HFpEF and HFmrEF So Different? The Need to Understand Distinct Phenotypes. Front Cardiovasc Med. 2021, 8, 676658. [Google Scholar] [CrossRef]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.L., Jr.; Kiernan, M.S.; et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e1054–e1091, Erratum in Circulation 2017, 136, e345. [Google Scholar] [CrossRef]
- Kociol, R.D.; Pang, P.S.; Gheorghiade, M.; Fonarow, G.C.; O’Connor, C.M.; Felker, G.M. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J. Am. Coll. Cardiol. 2010, 56, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, B. Heart failure preserved ejection fraction with coronary artery disease: Time for a new classification? J. Am. Coll. Cardiol. 2014, 63 Pt A, 2828–2830. [Google Scholar] [CrossRef] [Green Version]
- Obokata, M.; Reddy, Y.N.V.; Melenovsky, V.; Kane, G.C.; Olson, T.P.; Jarolim, P.; Borlaug, B.A. Myocardial Injury and Cardiac Reserve in Patients with Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2018, 72, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Gohar, A.; Chong, J.P.C.; Liew, O.W.; den Ruijter, H.; de Kleijn, D.P.V.; Sim, D.; Yeo, D.P.S.; Ong, H.Y.; Jaufeerally, F.; Leong, G.K.T.; et al. The prognostic value of highly sensitive cardiac troponin assays for adverse events in men and women with stable heart failure and a preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1638–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myhre, P.L.; O’Meara, E.; Claggett, B.L.; de Denus, S.; Jarolim, P.; Anand, I.S.; Beldhuis, I.E.; Fleg, J.L.; Lewis, E.; Pitt, B.; et al. Cardiac Troponin I and Risk of Cardiac Events in Patients with Heart Failure and Preserved Ejection Fraction. Circ. Heart Fail. 2018, 11, e005312. [Google Scholar] [CrossRef]
- Gori, M.; Senni, M.; Claggett, B.; Liu, J.; Maggioni, A.P.; Zile, M.; Prescott, M.F.; Van Veldhuisen, D.J.; Zannad, F.; Pieske, B.; et al. Integrating High-Sensitivity Troponin T and Sacubitril/Valsartan Treatment in HFpEF: The PARAGON-HF Trial. JACC Heart Fail. 2021, 9, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Bayes-Genis, A.; Mebazaa, A.; Bauersachs, J.; Cleland, J.G.F.; Coats, A.J.S.; Januzzi, J.L.; Maisel, A.S.; McDonald, K.; Mueller, T.; et al. Circulating heart failure biomarkers beyond natriuretic peptides: Review from the Biomarker Study Group of the Heart Failure Association (HFA), European Society of Cardiology (ESC). Eur. J. Heart Fail. 2021, 23, 1610–1632. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.; Mueller, C.; Nowak, R.; Peacock, W.F.; Landsberg, J.W.; Ponikowski, P.; Mockel, M.; Hogan, C.; Wu, A.H.; Richards, M.; et al. Mid-region pro-hormone markers for diagnosis and prognosis in acute dyspnea: Results from the BACH (Biomarkers in Acute Heart Failure) trial. J. Am. Coll. Cardiol. 2010, 55, 2062–2076. [Google Scholar] [CrossRef] [Green Version]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Tromp, J.; Khan, M.A.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients with Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef]
- Sakane, K.; Kanzaki, Y.; Tsuda, K.; Maeda, D.; Sohmiya, K.; Hoshiga, M. Disproportionately low BNP levels in patients of acute heart failure with preserved vs. reduced ejection fraction. Int. J. Cardiol. 2021, 327, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Remmelzwaal, S.; van Ballegooijen, A.J.; Schoonmade, L.J.; Dal Canto, E.; Handoko, M.L.; Henkens, M.T.H.M.; van Empel, V.; Heymans, S.R.B.; Beulens, J.W.J. Natriuretic peptides for the detection of diastolic dysfunction and heart failure with preserved ejection fraction-a systematic review and meta-analysis. BMC Med. 2020, 18, 290. [Google Scholar] [CrossRef] [PubMed]
- Lopuszynski, J.B.; Downing, A.J.; Finley, C.M.; Zahid, M. Prognosticators of All-Cause Mortality in Patients with Heart Failure With Preserved Ejection Fraction. Am. J. Cardiol. 2021, 158, 66–73. [Google Scholar] [CrossRef]
- Kociol, R.D.; Horton, J.R.; Fonarow, G.C.; Reyes, E.M.; Shaw, L.K.; O’Connor, C.M.; Felker, G.M.; Hernandez, A.F. Admission, discharge, or change in B-type natriuretic peptide and long-term outcomes: Data from Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) linked to Medicare claims. Circ. Heart Fail. 2011, 4, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikimi, T.; Nakagawa, Y. Adrenomedullin as a Biomarker of Heart Failure. Heart Fail. Clin. 2018, 14, 49–55. [Google Scholar] [CrossRef]
- Voors, A.A.; Kremer, D.; Geven, C.; Ter Maaten, J.M.; Struck, J.; Bergmann, A.; Pickkers, P.; Metra, M.; Mebazaa, A.; Düngen, H.D.; et al. Adrenomedullin in heart failure: Pathophysiology and therapeutic application. Eur. J. Heart Fail. 2019, 21, 163–171. [Google Scholar] [CrossRef]
- Kremer, D.; Ter Maaten, J.M.; Voors, A.A. Bio-adrenomedullin as a potential quick, reliable, and objective marker of congestion in heart failure. Eur. J. Heart Fail. 2018, 20, 1363–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Maaten, J.M.; Kremer, D.; Demissei, B.G.; Struck, J.; Bergmann, A.; Anker, S.D.; Ng, L.L.; Dickstein, K.; Metra, M.; Samani, N.J.; et al. Bio-adrenomedullin as a marker of congestion in patients with new-onset and worsening heart failure. Eur. J. Heart Fail. 2019, 21, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Kozhuharov, N.; Ng, L.; Wussler, D.; Strebel, I.; Sabti, Z.; Hartmann, O.; Eltayeb, M.; Squire, I.; Nowak, A.; Rieger, M.; et al. Activity of the adrenomedullin system to personalise post-discharge diuretic treatment in acute heart failure. Clin Res Cardiol. 2022, 111, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Pandhi, P.; Ter Maaten, J.M.; Emmens, J.E.; Struck, J.; Bergmann, A.; Cleland, J.G.; Givertz, M.M.; Metra, M.; O’Connor, C.M.; Teerlink, J.R.; et al. Clinical value of pre-discharge bio-adrenomedullin as a marker of residual congestion and high risk of heart failure hospital readmission. Eur. J. Heart Fail. 2020, 22, 683–691. [Google Scholar] [CrossRef]
- Sharma, U.C.; Pokharel, S.; van Brakel, T.J.; van Berlo, J.H.; Cleutjens, J.P.; Schroen, B.; André, S.; Crijns, H.J.; Gabius, H.J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef]
- de Boer, R.A.; Voors, A.A.; Muntendam, P.; van Gilst, W.H.; van Veldhuisen, D.J. Galectin-3: A novel mediator of heart failure development and progression. Eur. J. Heart Fail. 2009, 11, 811–817. [Google Scholar] [CrossRef]
- Beltrami, M.; Ruocco, G.; Dastidar, A.G.; Franci, B.; Lucani, B.; Aloia, E.; Nuti, R.; Palazzuoli, A. Additional value of Galectin-3 to BNP in acute heart failure patients with preserved ejection fraction. Clin Chim Acta 2016, 457, 99–105. [Google Scholar] [CrossRef] [Green Version]
- de Boer, R.A.; Lok, D.J.; Jaarsma, T.; van der Meer, P.; Voors, A.A.; Hillege, H.L.; van Veldhuisen, D.J. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann. Med. 2011, 43, 60–68. [Google Scholar] [CrossRef]
- Edelmann, F.; Holzendorf, V.; Wachter, R.; Nolte, K.; Schmidt, A.G.; Kraigher-Krainer, E.; Duvinage, A.; Unkelbach, I.; Düngen, H.D.; Tschöpe, C.; et al. Galectin-3 in patients with heart failure with preserved ejection fraction: Results from the Aldo-DHF trial. Eur. J. Heart Fail. 2015, 17, 214–223. [Google Scholar] [CrossRef]
- Ghorbani, A.; Bhambhani, V.; Christenson, R.H.; Meijers, W.C.; de Boer, R.A.; Levy, D.; Larson, M.G.; Ho, J.E. Longitudinal Change in Galectin-3 and Incident Cardiovascular Outcomes. J. Am. Coll. Cardiol. 2018, 72, 3246–3254. [Google Scholar] [CrossRef]
- Weinberg, E.O.; Shimpo, M.; De Keulenaer, G.W.; MacGillivray, C.; Tominaga, S.; Solomon, S.D.; Rouleau, J.L.; Lee, R.T. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation 2002, 106, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Ky, B.; French, B.; McCloskey, K.; Rame, J.E.; McIntosh, E.; Shahi, P.; Dries, D.L.; Tang, W.H.; Wu, A.H.; Fang, J.C.; et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ. Heart Fail. 2011, 4, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Yu, C.C.; Chiu, F.C.; Tsai, C.T.; Lai, L.P.; Hwang, J.J.; Lin, J.L. Soluble ST2 as a biomarker for detecting stable heart failure with a normal ejection fraction in hypertensive patients. J. Card. Fail. 2013, 19, 163–168. [Google Scholar] [CrossRef]
- Aimo, A.; Vergaro, G.; Ripoli, A.; Bayes-Genis, A.; Pascual Figal, D.A.; de Boer, R.A.; Lassus, J.; Mebazaa, A.; Gayat, E.; Breidthardt, T.; et al. Meta-Analysis of Soluble Suppression of Tumorigenicity-2 and Prognosis in Acute Heart Failure. JACC Heart Fail. 2017, 5, 287–296. [Google Scholar] [CrossRef]
- Krebber, M.M.; van Dijk, C.G.M.; Vernooij, R.W.M.; Brandt, M.M.; Emter, C.A.; Rau, C.D.; Fledderus, J.O.; Duncker, D.J.; Verhaar, M.C.; Cheng, C.; et al. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Extracellular Matrix Remodeling during Left Ventricular Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 6742. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.M.; Ferreira, S.M.; Ferreira, M.J.; Falcão-Pires, I. Circulating Biomarkers of Collagen Metabolism and Prognosis of Heart Failure with Reduced or Mid-Range Ejection Fraction. Curr. Pharm. Des. 2017, 23, 3217–3223. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.W.; Claggett, B.L.; O’Meara, E.; Prescott, M.F.; Pfeffer, M.A.; Shah, S.J.; Redfield, M.M.; Zannad, F.; Chiang, L.M.; Rizkala, A.R.; et al. Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients with HFpEF. J. Am. Coll. Cardiol. 2020, 76, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Duprez, D.A.; Gross, M.D.; Kizer, J.R.; Ix, J.H.; Hundley, W.G.; Jacobs, D.R., Jr. Predictive Value of Collagen Biomarkers for Heart Failure with and without Preserved Ejection Fraction: MESA (Multi-Ethnic Study of Atherosclerosis). J. Am. Heart Assoc. 2018, 7, e007885. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Benjamin, E.J.; Siwik, D.A.; Safa, R.; Guo, C.Y.; Corey, D.; Sundstrom, J.; Sawyer, D.B.; Colucci, W.S.; et al. Clinical and echocardiographic correlates of plasma procollagen type III amino-terminal peptide levels in the community. Am. Heart J. 2007, 154, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, I.; Glazer, N.L.; Barasch, E.; Djousse, L.; Gottdiener, J.S.; Ix, J.H.; Kizer, J.R.; Rimm, E.B.; Siscovick, D.S.; King, G.L.; et al. Associations between metabolic dysregulation and circulating biomarkers of fibrosis: The Cardiovascular Health Study. Metabolism 2015, 64, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasceri, V.; Willerson, J.T.; Yeh, E.T. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation 2000, 102, 2165–2168. [Google Scholar] [CrossRef]
- Koller, L.; Kleber, M.; Goliasch, G.; Sulzgruber, P.; Scharnagl, H.; Silbernagel, G.; Grammer, T.; Delgado, G.; Tomaschitz, A.; Pilz, S.; et al. C-reactive protein predicts mortality in patients referred for coronary angiography and symptoms of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2014, 16, 758–766. [Google Scholar] [CrossRef]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e007385. [Google Scholar] [CrossRef]
- DuBrock, H.M.; AbouEzzeddine, O.F.; Redfield, M.M. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS ONE 2018, 13, e0201836. [Google Scholar] [CrossRef]
- Kempf, T.; von Haehling, S.; Peter, T.; Allhoff, T.; Cicoira, M.; Doehner, W.; Ponikowski, P.; Filippatos, G.S.; Rozentryt, P.; Drexler, H.; et al. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.M.; Santhanakrishnan, R.; Chong, J.P.; Chen, Z.; Tai, B.C.; Liew, O.W.; Ng, T.P.; Ling, L.H.; Sim, D.; Leong, K.T.G.; et al. Growth differentiation factor 15 in heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 2016, 18, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Kato, E.T.; Morrow, D.A.; Guo, J.; Berg, D.D.; Blazing, M.A.; Bohula, E.A.; Bonaca, M.P.; Cannon, C.P.; de Lemos, J.A.; Giugliano, R.P.; et al. Growth differentiation factor 15 and cardiovascular risk: Individual patient meta-analysis. Eur. Heart J. 2022, ehac577. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Chia, Y.C. Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J. Am. Heart Assoc. 2021, 10, e018549. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.C.; Gan, L.M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure With Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10, e001633. [Google Scholar] [CrossRef] [Green Version]
- Marti, C.N.; Khan, H.; Mann, D.L.; Georgiopoulou, V.V.; Bibbins-Domingo, K.; Harris, T.; Koster, A.; Newman, A.; Kritchevsky, S.B.; Kalogeropoulos, A.P.; et al. Soluble tumor necrosis factor receptors and heart failure risk in older adults: Health, Aging, and Body Composition (Health ABC) Study. Circ. Heart Fail. 2014, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Shantsila, E.; Wrigley, B.J.; Blann, A.D.; Gill, P.S.; Lip, G.Y. A contemporary view on endothelial function in heart failure. Eur. J. Heart Fail. 2012, 14, 873–881. [Google Scholar] [CrossRef]
- Shah, S.J.; Lam, C.S.P.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.S.; Beussink-Nelson, L.; Ljung Faxén, U.; Fermer, M.L.; Broberg, M.A.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450, Erratum in Eur. Heart J. 2019, 40, 541.. [Google Scholar] [CrossRef]
- Rush, C.J.; Berry, C.; Oldroyd, K.G.; Rocchiccioli, J.P.; Lindsay, M.M.; Touyz, R.M.; Murphy, C.L.; Ford, T.J.; Sidik, N.; McEntegart, M.B.; et al. Prevalence of Coronary Artery Disease and Coronary Microvascular Dysfunction in Patients with Heart Failure with Preserved Ejection Fraction. JAMA Cardiol. 2021, 6, 1130–1143. [Google Scholar] [CrossRef]
- Emdin, M.; Aimo, A.; Castiglione, V.; Vergaro, G.; Georgiopoulos, G.; Saccaro, L.F.; Lombardi, C.M.; Passino, C.; Cerbai, E.; Metra, M.; et al. Targeting Cyclic Guanosine Monophosphate to Treat Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 1795–1807. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Role of endothelin-1 in hypertension and vascular disease. Am. J. Hypertens. 2001, 14 Pt 2, 83S–89S. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, M.A.; Moukarbel, G.V.; Gupta, R.; Frank, S.M.; Anderson, A.M.; Liu, L.C.; Khouri, S.J. Endothelin 1 Is Associated with Heart Failure Hospitalization and Long-Term Mortality in Patients with Heart Failure with Preserved Ejection Fraction and Pulmonary Hypertension. Cardiology 2019, 143, 124–133. [Google Scholar] [CrossRef]
- Bevan, G.H.; Jenkins, T.; Josephson, R.; Rajagopalan, S.; Al-Kindi, S.G. Endothelin-1 and peak oxygen consumption in patients with heart failure with preserved ejection fraction. Heart Lung 2021, 50, 442–446. [Google Scholar] [CrossRef]
- Winter, M.P.; Kleber, M.E.; Koller, L.; Sulzgruber, P.; Scharnagl, H.; Delgado, G.; Goliasch, G.; März, W.; Niessner, A. Prognostic significance of tPA/PAI-1 complex in patients with heart failure and preserved ejection fraction. Thromb. Haemost. 2017, 117, 471–478. [Google Scholar] [CrossRef]
- Jug, B.; Vene, N.; Salobir, B.G.; Sebestjen, M.; Sabovic, M.; Keber, I. Procoagulant state in heart failure with preserved left ventricular ejection fraction. Int. Heart J. 2009, 50, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Hage, C.; Bjerre, M.; Frystyk, J.; Gu, H.F.; Brismar, K.; Donal, E.; Daubert, J.C.; Linde, C.; Lund, L.H. Comparison of Prognostic Usefulness of Serum Insulin-Like Growth Factor-Binding Protein 7 in Patients with Heart Failure and Preserved versus Reduced Left Ventricular Ejection Fraction. Am. J. Cardiol. 2018, 121, 1558–1566. [Google Scholar] [CrossRef]
- Sabbah, M.S.; Fayyaz, A.U.; de Denus, S.; Felker, G.M.; Borlaug, B.A.; Dasari, S.; Carter, R.E.; Redfield, M.M. Obese-Inflammatory Phenotypes in Heart Failure with Preserved Ejection Fraction. Circ. Heart Fail. 2020, 13, e006414. [Google Scholar] [CrossRef]
- Gandhi, P.U.; Chow, S.L.; Rector, T.S.; Krum, H.; Gaggin, H.K.; McMurray, J.J.; Zile, M.R.; Komajda, M.; McKelvie, R.S.; Carson, P.E.; et al. Prognostic Value of Insulin-Like Growth Factor-Binding Protein 7 in Patients with Heart Failure and Preserved Ejection Fraction. J. Card. Fail. 2017, 23, 20–28. [Google Scholar] [CrossRef]
- Gandhi, P.U.; Gaggin, H.K.; Redfield, M.M.; Chen, H.H.; Stevens, S.R.; Anstrom, K.J.; Semigran, M.J.; Liu, P.; Januzzi, J.L., Jr. Insulin-Like Growth Factor-Binding Protein-7 as a Biomarker of Diastolic Dysfunction and Functional Capacity in Heart Failure with Preserved Ejection Fraction: Results from the RELAX Trial. JACC Heart Fail. 2016, 4, 860–869. [Google Scholar] [CrossRef]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure with Preserved Ejection Fraction and Heart Failure with Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef]
- Ferro, F.; Spelat, R.; Valente, C.; Contessotto, P. Understanding How Heart Metabolic Derangement Shows Differential Stage Specificity for Heart Failure with Preserved and Reduced Ejection Fraction. Biomolecules 2022, 12, 969. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Cediel, G.; Domingo, M.; Codina, P.; Santiago, E.; Lupón, J. Biomarkers in Heart Failure with Preserved Ejection Fraction. Card. Fail. Rev. 2022, 8, e20. [Google Scholar] [CrossRef]
- Zhou, X.; He, L.; Zuo, S.; Zhang, Y.; Wan, D.; Long, C.; Huang, P.; Wu, X.; Wu, C.; Liu, G.; et al. Serine prevented high-fat diet-induced oxidative stress by activating AMPK and epigenetically modulating the expression of glutathione synthesis-related genes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 488–498. [Google Scholar] [CrossRef]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Puckett, D.L.; Alquraishi, M.; Chowanadisai, W.; Bettaieb, A. The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 1171. [Google Scholar] [CrossRef]
- Chen, S.; Xia, Y.; He, F.; Fu, J.; Xin, Z.; Deng, B.; He, L.; Zhou, X.; Ren, W. Serine Supports IL-1β Production in Macrophages through mTOR Signaling. Front. Immunol. Sec. Nutr. Immunol. 2020. [Google Scholar] [CrossRef]
- Fu, Z.; Akula, S.; Thorpe, M.; Hellman, L. Potent and Broad but not Unselective Cleavage of Cytokines and Chemokines by Human Neutrophil Elastase and Proteinase 3. Int. J. Mol. Sci. 2020, 21, 651. [Google Scholar] [CrossRef]
- Lejeune, S.; Menghoum, N.; Thompson, J.; Robillard, I.; Hussin, J.; Bertrand, L.; Horman, S.; Des Rosiers, C.; Beauloye, C.; Pouleur, A.-C. Plasma metabolomics identify hydroxyproline as a potential player in the pathophysiogy of HFpEF. Arch. Cardiovasc. Dis.Suppl. 2022, 14, 193. [Google Scholar] [CrossRef]
- Chen, F.; Lucas, R.; Fulton, D. The subcellular compartmentalization of arginine metabolizing enzymes and their role in endothelial dysfunction. Front. Immunol. 2013, 4, 184. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, J.; Khondkar, W.; Morelli, M.B.; Wang, X.; Santulli, G.; Trimarco, V. Arginine and Endothelial Function. Biomedicines 2020, 8, 277. [Google Scholar] [CrossRef]
- Tang, W.H.; Tong, W.; Shrestha, K.; Wang, Z.; Levison, B.S.; Delfraino, B.; Hu, B.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Differential effects of arginine methylation on diastolic dysfunction and disease progression in patients with chronic systolic heart failure. Eur. Heart J. 2008, 29, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol.Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Wu, C.; Xu, Y.; Cai, J.; Zhao, M.; Zu, L. The NO-cGMP-PKG Axis in HFpEF: From Pathological Mechanisms to Potential Therapies. Aging Dis. 2022. [Google Scholar] [CrossRef]
- Phillips, C.M.; Chen, L.W.; Heude, B.; Bernard, J.Y.; Harvey, N.C.; Duijts, L.; Mensink-Bout, S.M.; Polanska, K.; Mancano, G.; Suderman, M.; et al. Dietary Inflammatory Index and Non-Communicable Disease Risk: A Narrative Review. Nutrients 2019, 11, 1873. [Google Scholar] [CrossRef] [Green Version]
- Bak, D.W.; Weerapana, E. Cysteine-mediated redox signalling in the mitochondria. Mol. Biosyst. 2015, 11, 678–697. [Google Scholar] [CrossRef]
- Zweier, J.L.; Talukder, M.A.H. The role of oxidants and free radicals in reperfusion injury. Cardiovasc. Res. 2006, 70, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Bekfani, T.; Bekhite, M.; Neugebauer, S.; Derlien, S.; Hamadanchi, A.; Nisser, J.; Hilse, M.S.; Haase, D.; Kretzschmar, T.; Wu, M.F.; et al. Metabolomic Profiling in Patients with Heart Failure and Exercise Intolerance: Kynurenine as a Potential Biomarker. Cells 2022, 11, 1674. [Google Scholar] [CrossRef]
- De Jong, K.A.; Nikolaev, V.O. Multifaceted remodelling of cAMP microdomains driven by different aetiologies of heart failure. FEBS J. 2021, 288, 6603–6622. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef]
- Zordoky, B.N.; Sung, M.M.; Ezekowitz, J.; Mandal, R.; Han, B.; Bjorndahl, T.C.; Bouatra, S.; Anderson, T.; Oudit, G.Y.; Wishart, D.S.; et al. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS ONE 2015, 10, e0124844. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Ferro, F.; Ouillé, A.; Tran, T.A.; Fontanaud, P.; Bois, P.; Babuty, D.; Labarthe, F.; Le Guennec, J.Y. Long-chain acylcarnitines regulate the hERG channel. PLoS ONE 2012, 7, e41686. [Google Scholar] [CrossRef] [Green Version]
- Trovato, F.; Zia, R.; Napoli, S.; Wolfer, K.; Huang, X.; Morgan, P.E.; Husbyn, H.; Elgosbi, M.; Lucangeli, M.; Miquel, R.; et al. Dysregulation of the Lysophosphatidylcholine/Autotaxin/Lysophosphatidic Acid Axis in Acute-on-Chronic Liver Failure Is Associated with Mortality and Systemic Inflammation by Lysophosphatidic Acid-Dependent Monocyte Activation. Hepatology 2021, 74, 907–925. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Vegter, E.L.; van der Meer, P.; de Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Du, T.; Long, T.; Liao, X.; Dong, Y.; Huang, Z.-P. Signaling cascades in the failing heart and emerging therapeutic strategies. Signal Transduct. Target. Ther. 2022, 7, 134. [Google Scholar] [CrossRef]
- Wong, L.L.; Zou, R.; Zhou, L.; Lim, J.Y.; Phua, D.C.Y.; Liu, C.; Chong, J.P.C.; Ng, J.Y.X.; Liew, O.W.; Chan, S.P.; et al. Combining Circulating MicroRNA and NT-proBNP to Detect and Categorize Heart Failure Subtypes. J. Am. Coll. Cardiol. 2019, 73, 1300–1313. [Google Scholar] [CrossRef]
- Chen, F.; Yang, J.; Li, Y.; Wang, H. Circulating microRNAs as novel biomarkers for heart failure. Hell. J.Cardiol. 2018, 59, 209–214. [Google Scholar] [CrossRef]
- Sucharov, C.C.; Kao, D.P.; Port, J.D.; Karimpour-Fard, A.; Quaife, R.A.; Minobe, W.; Nunley, K.; Lowes, B.D.; Gilbert, E.M.; Bristow, M.R. Myocardial microRNAs associated with reverse remodeling in human heart failure. JCI Insight 2017, 2, e89169. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name of Biomarker | Mechanism of Action | |
|---|---|---|
| Markers of myocardial injury | ↑↑ High sensitivity troponin | The final results of microvascular dysfunction, and subendocardial layer damage due to systemic oxygen reduction. |
| ↑ Natriuretic peptides | Related to diuresis and natriuresis which favor congestion reduction and euvolemia. | |
| ↑↑ Adrenomedullin | Regulatory peptide produced by endothelial and smooth muscle cells with antiproliferative vasodilatatory and antiapoptotic effects. | |
| Markers of extracellular fibrosis | ↑↑ Galectin-3 | Inflammatory and pro-fibrotic processes |
| ↑↑ Soluble ST2 | Produced by myocardial cells, but smooth muscle cells and endothelium are also capable of synthesizing the peptide in relation to congestion. | |
| ↑↑ Matrix metalloproteinases | Involved in collagen synthesis and collagen degradation. | |
| ↑↑ Procollagen type I (PIP) and procollagen type III N-terminal peptide (PIINP) | Reflects collagen increase deposition and turnover. | |
| Markers of inflammation | ↑↑ CRP and pentraxin | Inducing complement and cytokine stimulation causing myocyte loss and endothelial dysfunction via NO production decrease. |
| ↑↑ Grow differentiation factor 15 | Expressed in inflammatory chronic diseases, lung, kidney, and cardiovascular diseases and providing additional information on LV remodeling and function. | |
| ↑↑ Intereleukin-6 | Contributes through direct myocyte damage and indirect inflammatory burden elevation. | |
| ↑ Tumor necrosis factor α | Correlates with atrial dimension and diastolic dysfunction degree. | |
| Markers of endothelial dysfunction | ↑↑ Vascular cell adhesion molecules (VCAM) and E selectin ↑ Endothelin 1 | Activates von Willebrand and other prothrombotic factors secreted by the endothelial cells in response to renin angiotensin system activation. |
| ↑↑ Plasminogen activator inhibitor ↑↑ Insulin grow factor binding | In association with D-dimer levels suggesting an association with prothrombotic and procoagulant state.Left atrial dysfunction and dilatation reflecting diastolic dysfunction in HFpEF. |
| Biomarkers | Altered Cell Mechanism | |
|---|---|---|
| Increased inflammation | ↓ Serine ↑ Cathepsin G | Immunoregulatory actions: essential for production of proinflammatory cytokines in M1 macrophages stimulating the production of cytokines and chemokines. |
| ↑ Cystine | Key player in conditions of oxidative stress. | |
| ↑ Kynurenine | Controls local and systemic immune responses. | |
| Increased collagen synthesis and reduced myocardial compliance | ↑ Hydroxyproline | Role of stability of collagen and this dysregulation contributes to myocardial fibrosis. |
| ↑ Elastase | Degradation of extracellular matrix components, including collagen, elastin and fibronectin. | |
| ↓ cGMP/PKG signaling | Phosphorylation reduction associated with passive stiffness of cardiac muscle. | |
| Endothelial dysfunction | ↓ Arginine | Substrate for NO production by endothelial cells with reduced vasodilatory effects. |
| ↑ SDMA | Alternative methylation product of L-arginine associated with worsening renal function and microvascular dysfunction. | |
| Energetic impairment | ↓ cAMP | Is produced via β-AR signaling. |
| ↑ Acylcarnitine | Implies inefficient β-oxidation. | |
| ↑ Tryptophan | Produces metabolites including kynurenic acid and nicotinamide adenine dinucleotide. | |
| Metabolic lipid impairment | ↓ Lysophosphatidylcholine | Is required for the assembly of VLDLs and chylomicrons. |
| ↓ cAMP | Involved in lipolysis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palazzuoli, A.; Tramonte, F.; Beltrami, M. Laboratory and Metabolomic Fingerprint in Heart Failure with Preserved Ejection Fraction: From Clinical Classification to Biomarker Signature. Biomolecules 2023, 13, 173. https://doi.org/10.3390/biom13010173
Palazzuoli A, Tramonte F, Beltrami M. Laboratory and Metabolomic Fingerprint in Heart Failure with Preserved Ejection Fraction: From Clinical Classification to Biomarker Signature. Biomolecules. 2023; 13(1):173. https://doi.org/10.3390/biom13010173
Chicago/Turabian StylePalazzuoli, Alberto, Francesco Tramonte, and Matteo Beltrami. 2023. "Laboratory and Metabolomic Fingerprint in Heart Failure with Preserved Ejection Fraction: From Clinical Classification to Biomarker Signature" Biomolecules 13, no. 1: 173. https://doi.org/10.3390/biom13010173
APA StylePalazzuoli, A., Tramonte, F., & Beltrami, M. (2023). Laboratory and Metabolomic Fingerprint in Heart Failure with Preserved Ejection Fraction: From Clinical Classification to Biomarker Signature. Biomolecules, 13(1), 173. https://doi.org/10.3390/biom13010173