
Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors
 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
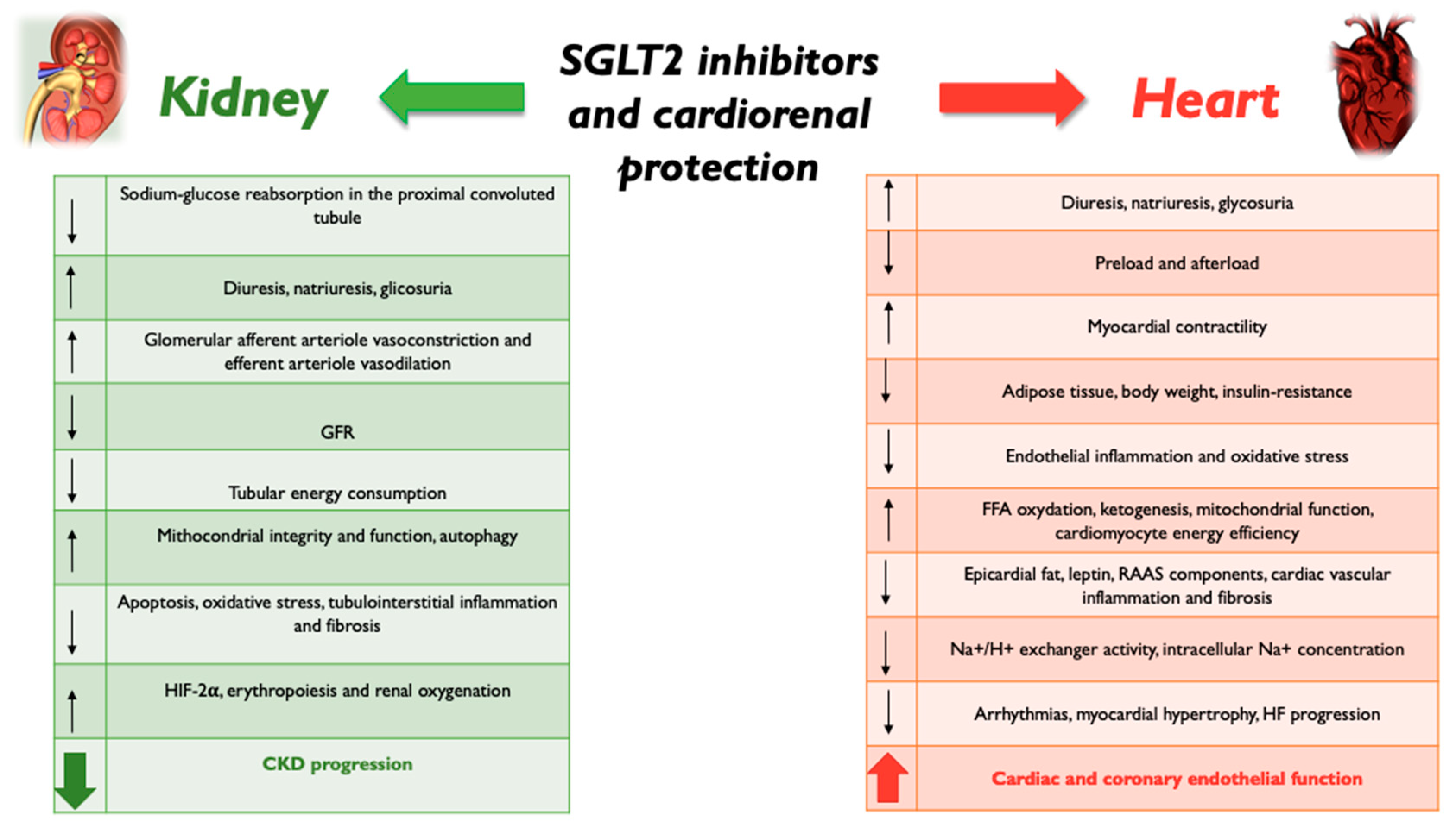
2. SGLT2 Inhibition and Kidney Protection
3. SGLT2 Inhibition and Heart Protection
4. Changes in Biomarkers Linked with Diabetic Kidney Disease and Heart Failure with SGLT2-Is
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.N.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.J.; Boyko, E.J.; Balkau, B.; Barengo, N.; Barr, E.; Basit, A.; Bhata, D.; Bommer, C.; Booth, G.; Cariou, B.; et al. International Diabetes Federation Diabetes Atlas, 10th ed.; IDF: Brussels, Belgium, 2021; Available online: https://www.diabetesatlas.org/en (accessed on 15 April 2022).
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Prandi, F.R.; Lecis, D.; Illuminato, F.; Milite, M.; Celotto, R.; Lerakis, S.; Romeo, F.; Barillà, F. Epigenetic Modifications and Non-Coding RNA in Diabetes-Mellitus-Induced Coronary Artery Disease: Pathophysiological Link and New Therapeutic Frontiers. Int. J. Mol. Sci. 2022, 23, 4589. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991, 14, 173–194. [Google Scholar] [CrossRef]
- Wagenmakers, A.J. Insulin resistance in the offspring of parents with type 2 diabetes. PLoS Med. 2005, 2, e289. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef]
- Cerasi, E.; Luft, R.; Efendic, S. Decreased Sensitivity of the Pancreatic Beta Cells to Glucose in Prediabetic and Diabetic Subjects: A Glucose Dose-Response Study. Diabetes 1972, 21, 224–234. [Google Scholar] [CrossRef]
- Holman, N.; Young, B.; Gadsby, R. Current prevalence of type 1 and type 2 diabetes in adults and children in the UK. Diabetes Med. 2015, 32, 1119–1120. [Google Scholar] [CrossRef]
- Cornel, J.H.; Bakris, G.L.; Stevens, S.R.; Alvarsson, M.; Bax, W.A.; Chuang, L.M.; Engel, S.S.; Lopes, R.D.; McGuire, D.K.; Riefflin, A.; et al. Effect of sitagliptin on kidney function and respective cardiovascular outcomes in type 2 diabetes: Outcomes from TECOS. Diabetes Care 2016, 39, 2304–2310. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Scirica, B.M.; Braunwald, E.; Raz, I.; Cavender, M.A.; Morrow, D.A.; Jarolim, P.; Udell, J.; Mosenzon, O.; Im, K.; Umez-Eronini, A.A.; et al. Heart failure, saxagliptin, and diabetes mellitus: Observations from the SAVOR-TIMI 53 randomized trial. Circulation 2015, 132, e198. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.Z.; Mentz, R.J.; Stebbins, A.; Mikus, C.R.; Schulte, P.J.; Fleg, J.L.; Cooper, L.S.; Leifer, E.S.; Badenhop, D.T.; Keteyian, S.J.; et al. Response to exercise training and outcomes in patients with heart failure and diabetes mellitus: Insights from the HF-ACTION trial. J. Card. Fail. 2016, 22, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; O’Connor, C.; McMurray, J.J.V.; Wittes, J.; Abraham, W.T.; Anker, S.D.; Dickstein, K.; Filippatos, G.; Holcomb, R.; Krum, H.; et al. Effect of ularitide on cardiovascular mortality in acute heart failure. N. Engl. J. Med. 2017, 376, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, M.H.; Zuo, L.; Wang, Y.; Yu, F.; Zhang, H.; Wang, H. China Kidney Disease Network (CK-NET) 2015 annual data report. Kidney Int. Suppl. 2019, 9, e1–e81. [Google Scholar] [CrossRef]
- Rodriguez-Gutierrez, R.; Gonzalez-Gonzalez, J.G.; Zuniga-Hernandez, J.A.; McCoy, R.G. Benefits and harms of intensive glycemic control in patients with type 2 diabetes. BMJ 2019, 367, l5887. [Google Scholar] [CrossRef]
- Salvatore, T.; Carbonara, O.; Cozzolino, D.; Torella, R.; Nasti, R.; Lascar, N.; Sasso, F.C. Kidney in diabetes: From organ damage target to therapeutic target. Curr. Drug Metab. 2011, 12, 658–666. [Google Scholar] [CrossRef]
- Tsampasian, V.; Baral, R.; Chattopadhyay, R.; Debski, M.; Joshi, S.S.; Reinhold, J.; Dweck, M.R.; Garg, P.; Vassiliou, V.S. The Role of SGLT2 Inhibitors in Heart Failure: A Systematic Review and Meta-Analysis. Cardiol. Res. Pract. 2021, 19, 9927533. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef] [Green Version]
- Merovci, A.; Mari, A.; Solis-Herrera, C.; Xiong, J.; Daniele, G.; Chavez-Velazquez, A.; Tripathy, D.; Urban McCarthy, S.; Abdul-Ghani, M.; DeFronzo, R.A. Dapagliflozin lowers plasma glucose concentration and improves β-cell function. J. Clin. Endocrinol. Metab. 2015, 100, 1927–1932, Erratum in J. Clin. Endocrinol. Metab. 2017, 102, 4662. [Google Scholar] [CrossRef] [PubMed]
- De Konink, L. Observations sur les proprietes febrifuges de la phloridzine. Bull. Soc. Med. Gand. 1836, 1, 75–110. [Google Scholar]
- Chasis, H.; Jolliffe, N.; Smith, H.W. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J. Clin. Investig. 1933, 12, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Fediuk, D.J.; Nucci, G.; Dawra, V.K.; Cutler, D.L.; Amin, N.B.; Terra, S.G.; Boyd, R.A.; Krishna, R.; Sahasrabudhe, V. Overview of the clinical pharmacology of ertugliflozin, a novel Sodium-Glucose Cotransporter 2 (SGLT2) inhibitor. Clin. Pharmacokinet. 2020, 59, 949–965. [Google Scholar] [CrossRef]
- Markham, A.; Elkinson, S. Luseogliflozin: First global approval. Drugs 2014, 74, 945–950. [Google Scholar] [CrossRef]
- Poole, R.M.; Prossler, J.E. Tofogliflozin: First global approval. Drugs 2014, 74, 39–44. [Google Scholar] [CrossRef]
- Caturano, A.; Galiero, R.; Pafundi, P.C.; Cesaro, A.; Vetrano, E.; Palmiero, G.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; et al. Does a strict glycemic control during acute coronary syndrome play a cardioprotective effect? Pathophysiology and clinical evidence. Diabetes Res. Clin. Pract. 2021, 178, 108959. [Google Scholar] [CrossRef]
- Kang, A.; Jardine, M.J. SGLT2 inhibitors may offer benefit beyond diabetes. Nat. Rev. Nephrol. 2021, 17, 83–84. [Google Scholar] [CrossRef]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. DAPA-HF trial Committees and Investigators. Dapaglifozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Keller, D.M.; Ahmed, N.; Tariq, H.; Walgamage, M.; Walgamage, T.; Mohammed, A.; Chou, J.T.; Kałużna-Oleksy, M.; Lesiak, M.; Straburzyńska-Migaj, E. SGLT2 Inhibitors in Type 2 Diabetes Mellitus and Heart Failure-A Concise Review. J. Clin. Med. 2022, 11, 1470. [Google Scholar] [CrossRef]
- Fitchett, D.; Inzucchi, S.E.; Cannon, C.P.; McGuire, D.K.; Scirica, B.M.; Johansen, O.E.; Sambevski, S.; Kaspers, S.; Pfarr, E.; George, J.T.; et al. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019, 139, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Madan Paramasivan, A.; Purushothaman, A.; Desouza, C. Implications of the CANVAS Study in Reducing Cardiovascular Outcomes. Curr. Diabetes Rep. 2018, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Sarraju, A.; Li, J.; Cannon, C.P.; Chang, T.I.; Agarwal, R.; Bakris, G.; Charytan, D.M.; de Zeeuw, D.; Greene, T.; Heerspink, H.J.L.; et al. Effects of canagliflozin on cardiovascular, renal, and safety outcomes in participants with type 2 diabetes and chronic kidney disease according to history of heart failure: Results from the CREDENCE trial. Am. Heart J. 2021, 233, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Mosenzon, O.; Wiviott, S.D.; Cahn, A.; Rozenberg, A.; Yanuv, I.; Goodrich, E.L.; Murphy, S.A.; Heerspink, H.J.L.; Zelniker, T.A.; Dwyer, J.P.; et al. Effects of dapagliflozin on development and progression of kidney disease in patients with type 2 diabetes: An analysis from the DECLARE-TIMI 58 randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 606–617. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Bonaca, M.P.; Furtado, R.H.M.; Mosenzon, O.; Kuder, J.F.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; McGuire, D.K.; Wilding, J.P.H.; et al. Effect of Dapagliflozin on Atrial Fibrillation in Patients with Type 2 Diabetes Mellitus: Insights From the DECLARE-TIMI 58 Trial. Circulation 2020, 141, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.V.; Jardine, M.J. SGLT2 inhibitors may prevent diabetes. Nat. Rev. Nephrol. 2022, 18, 203–204. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. DAPA-CKD Trial Committees and Investigators. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Ruhnau, B.; Faber, O.K.; Borch-Johnsen, K.; Thorsteinsson, B. Renal threshold for glucose in non-insulin-dependent diabetic patients. Diabetes Res. Clin. Pract. 1997, 36, 27–33. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; DeFronzo, R.A. Why do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in humans? Diabetes 2012, 61, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Braunwald, E. Cardiac and renal effects of sodium-glucose co-transporter 2 inhibitors in diabetes. J. Am. Coll. Cardiol. 2018, 72, 1845–1855. [Google Scholar] [CrossRef]
- Boyer, C.C. The vascular pattern of the renal glomerulus as revealed by plastic reconstruction from serial sections. Anat. Rec. 1956, 125, 433–441. [Google Scholar] [CrossRef]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef]
- Darshi, M.; Onishi, A.; Kim, J.J.; Pham, J.; Van Espen, B.F. Metabolic reprogramming in diabetic kidney disease can be restored via SGLT2 inhibition. J. Am. Soc. Nephrol. 2017, 28, 439. [Google Scholar]
- Mulder, S.; Heerspink, H.J.L.; Darshi, M.; Kim, J.J.; Laverman, G.D.; Sharma, K.; Pena, M.J. Effects of dapagliflozin on urinary metabolites in people with type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 2422–2428. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.-K.; Dong, Z. Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell. Mol. Life Sci. 2018, 75, 669–688. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Kurtz, A. Regulation of erythropoietin production. Eur. J. Clin. Investig. 2005, 35 (Suppl. S3), 13–19. [Google Scholar] [CrossRef] [PubMed]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann, S. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Langkilde, A.M.; Martinez, F.A.; Bengtsson, O.; Ponikowski, P.; Sabatine, M.S.; et al. DAPA-HF Committees and Investigators. A trial to evaluate the effect of the sodium-glucose co-transporter 2 inhibitor dapagliflozin on morbidity and mortality in patients with heart failure and reduced left ventricular ejection fraction (DAPA-HF). Eur. J. Heart Fail. 2019, 21, 665–675. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empaglifozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab. Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurry, J.J.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Verma, S. Potential mechanisms of sodium-glucose co-transporter 2 inhibitor-related cardiovascular benefits. Am. J. Med. 2019, 132, S39–S48. [Google Scholar] [CrossRef]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. 2014, 8, 262–275.e9. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.R.; Januszewicz, A.; Gilbert, R.E.; Vijapurkar, U.; Kline, I.; Fung, A.; Meininger, G. Effect of canagliflozin on blood pressure and adverse events related to osmotic diuresis and reduced intravascular volume in patients with type 2 diabetes mellitus. J. Clin. Hypertens. 2014, 16, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Polidori, D.; Heise, T.; Natarajan, J.; Farrell, K.; Wang, S.-S.; Sica, D.; Rothenberg, P.; Plum-Mörschel, L. Effect of the sodium glucose co-transporter 2 inhibitor canagliflozin on plasma volume in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2014, 16, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Kahl, S.; Gancheva, S.; Strabburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empaglifozin effectively lowers liver fat content in well controlled type 2 diabetes: A randomised, double blind, phase 4, placebo-controlled trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empaglifozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to fatty substrates utilization in response to sodium-glucose co-transporter2 inhibition in non-diabetic subjects and type 2 diabetic patients. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef]
- Ferrannini, F.; Mark, M.; Mayoux, E. CV protection in the EMPAREG OUTCOME trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Schulze, P.C.; Wu, J.M.F. Ketone bodies for the starving heart. Nat. Metab. 2020, 2, 1183–1185. [Google Scholar] [CrossRef]
- Challa, A.A.; Lewandowski, E.D. Short-Chain Carbon Sources: Exploiting Pleiotropic Effects for Heart Failure Therapy. JACC Basic Transl. Sci. 2022, 7, 730–742. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empaglifozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. College Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogwizd, S.M.; Sipido, K.R.; Verdonck, F.; Bers, D.M. Intracellular Na in animal models of hypertrophy and heart failure: Contractile function and arrhythmogenesis. Cardiovasc. Res. 2003, 57, 887–896. [Google Scholar] [CrossRef]
- Cingolani, H.E.; Ennis, I.L. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef]
- Leng, W.; Wu, M.; Pan, H.; Lei, X.; Chen, L.; Wu, Q.; Ouyang, X.; Liang, Z. The SGLT2 inhibitor dapaglifozin attenuates the activity of ROS-NLRP-3 inflammasome axis in steatohepatitis with diabetes mellitus. Ann. Transl. Med. 2019, 7, 429. [Google Scholar] [CrossRef]
- Papadopoulou-Marketou, N.; Kanaka-Gantenbein, C.; Marketos, N.; Chrousos, G.P.; Papassotiriou, I. Biomarkers of diabetic nephropathy: A 2017 update. Crit. Rev. Clin. Lab. Sci. 2017, 54, 326–342. [Google Scholar] [CrossRef]
- Michels, W.M.; Grootendorst, D.C.; Verduijn, M.; Elliott, E.G.; Dekker, F.W.; Krediet, R.T. Performance of the Cockcroft-Gault, MDRD, and new CKD-EPI formulas in relation to GFR, age, and body size. Clin. J. Am. Soc. Nephrol. 2010, 5, 1003–1009. [Google Scholar] [CrossRef]
- Krolewski, A.S. Progressive renal decline: The new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care 2015, 38, 954–962. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: Cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef]
- Levey, A.S.; Perrone, R.D.; Madias, N.E. Serum creatinine and renal function. Annu. Rev. Med. 1988, 39, 465–490. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, M.J.J.V.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; Cannon, C.P.; et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Furuki, T.; Kobori, H.; Miyakawa, M.; Imachi, H.; Murao, K.; Nishiyama, A. Effects of sodium-glucose cotransporter 2 inhibitors on urinary excretion of intact and total angiotensinogen in patients with type 2 diabetes. J. Investig. Med. 2017, 65, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L.; Butler, J.; Jarolim, P.; Sattar, N.; Vijapurkar, U.; Desai, M.; Davies, M.J. Effects of canagliflozin on cardiovascular biomarkers in older adults with type 2 diabetes. J. Am. Coll. Cardiol. 2017, 70, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Lessons learned from the DAPA-HF trial concerning the mechanisms of benefit of SGLT2 inhibitors on heart failure events in the context of other large-scale trials nearing completion. Cardiovasc. Diabetol. 2019, 18, 129. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Hatanaka, T.; Ogawa, D.; Tachibana, H.; Eguchi, J.; Inoue, T.; Yamada, H.; Akei, K.; Makino, H.; Wada, J. Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice. Pharmacol. Res. Perspect. 2016, 4, e00239. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 2018, 85, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prandi, F.R.; Barone, L.; Lecis, D.; Belli, M.; Sergi, D.; Milite, M.; Lerakis, S.; Romeo, F.; Barillà, F. Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors. Biomolecules 2022, 12, 1349. https://doi.org/10.3390/biom12101349
Prandi FR, Barone L, Lecis D, Belli M, Sergi D, Milite M, Lerakis S, Romeo F, Barillà F. Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors. Biomolecules. 2022; 12(10):1349. https://doi.org/10.3390/biom12101349
Chicago/Turabian StylePrandi, Francesca Romana, Lucy Barone, Dalgisio Lecis, Martina Belli, Domenico Sergi, Marialucia Milite, Stamatios Lerakis, Francesco Romeo, and Francesco Barillà. 2022. "Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors" Biomolecules 12, no. 10: 1349. https://doi.org/10.3390/biom12101349
APA StylePrandi, F. R., Barone, L., Lecis, D., Belli, M., Sergi, D., Milite, M., Lerakis, S., Romeo, F., & Barillà, F. (2022). Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors. Biomolecules, 12(10), 1349. https://doi.org/10.3390/biom12101349