
Tuning Liposome Stability in Biological Environments and Intracellular Drug Release Kinetics


Abstract
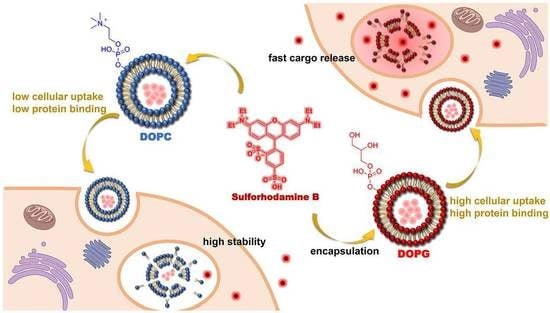
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Liposome Preparation and Characterization
2.2. Cell Culture
2.3. Flow Cytometry
2.4. Fluorescence Imaging
2.5. Size-Exclusion Chromatography
3. Results and Discussion
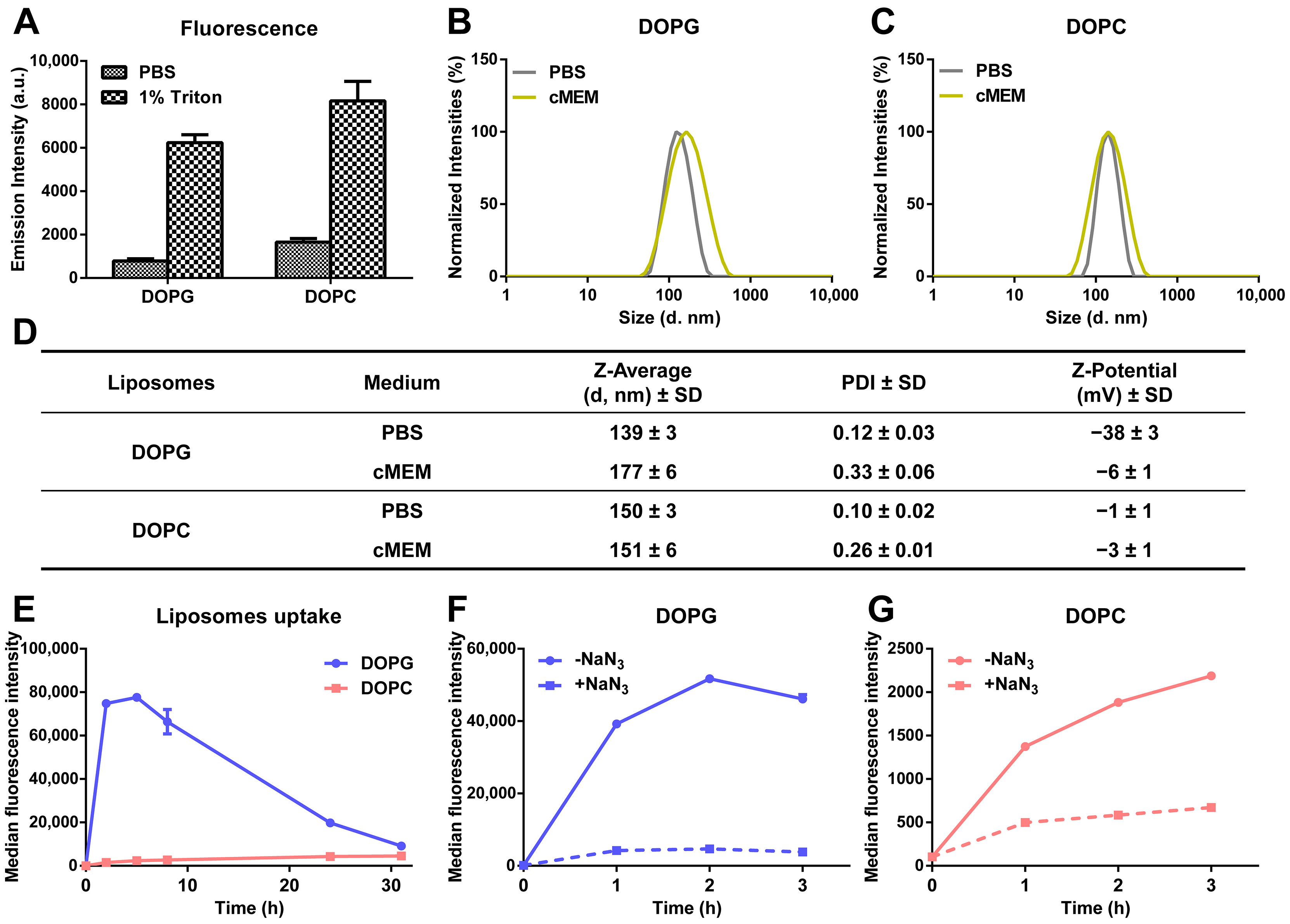
3.1. Liposome Preparation and Characterization
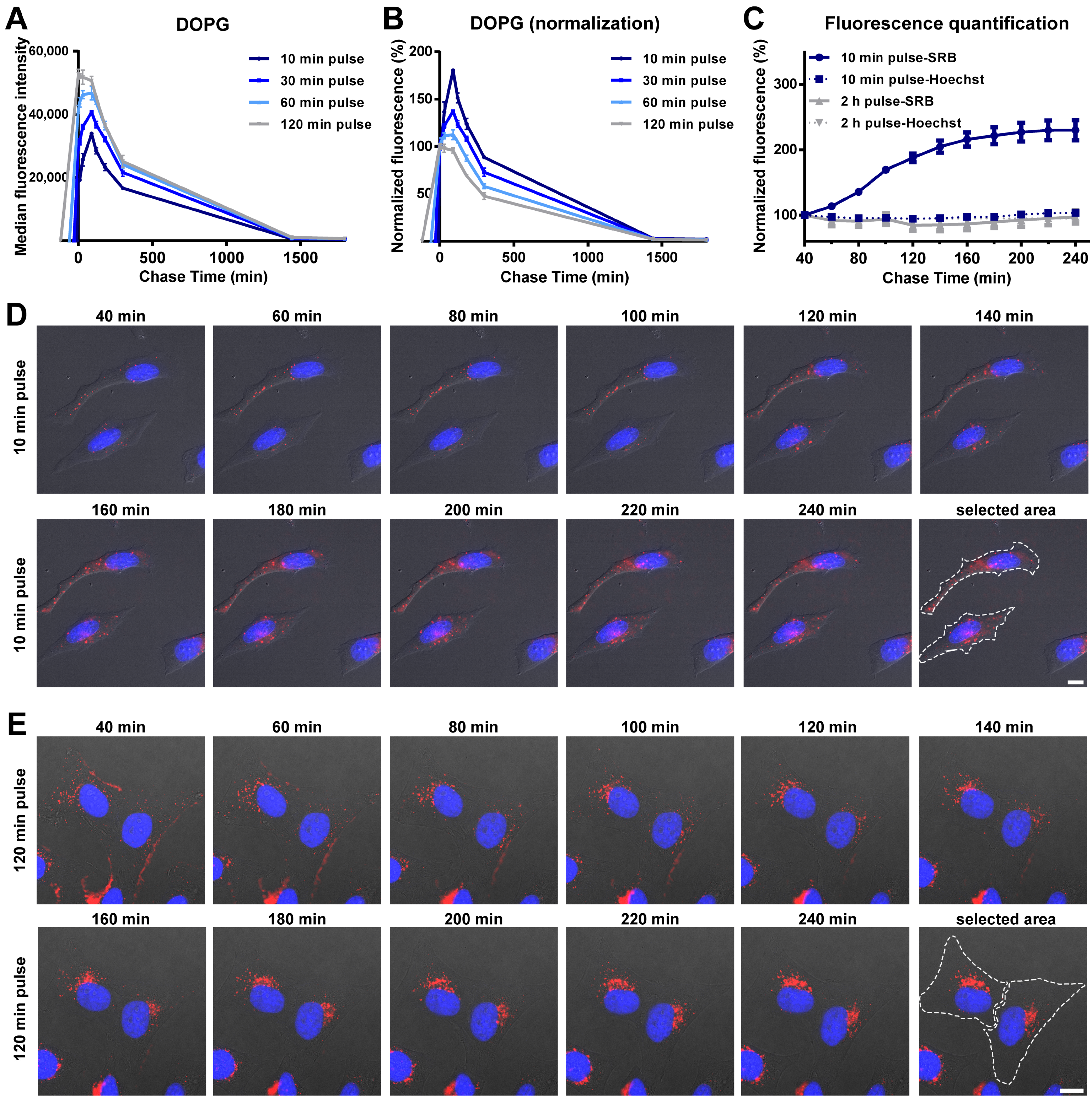
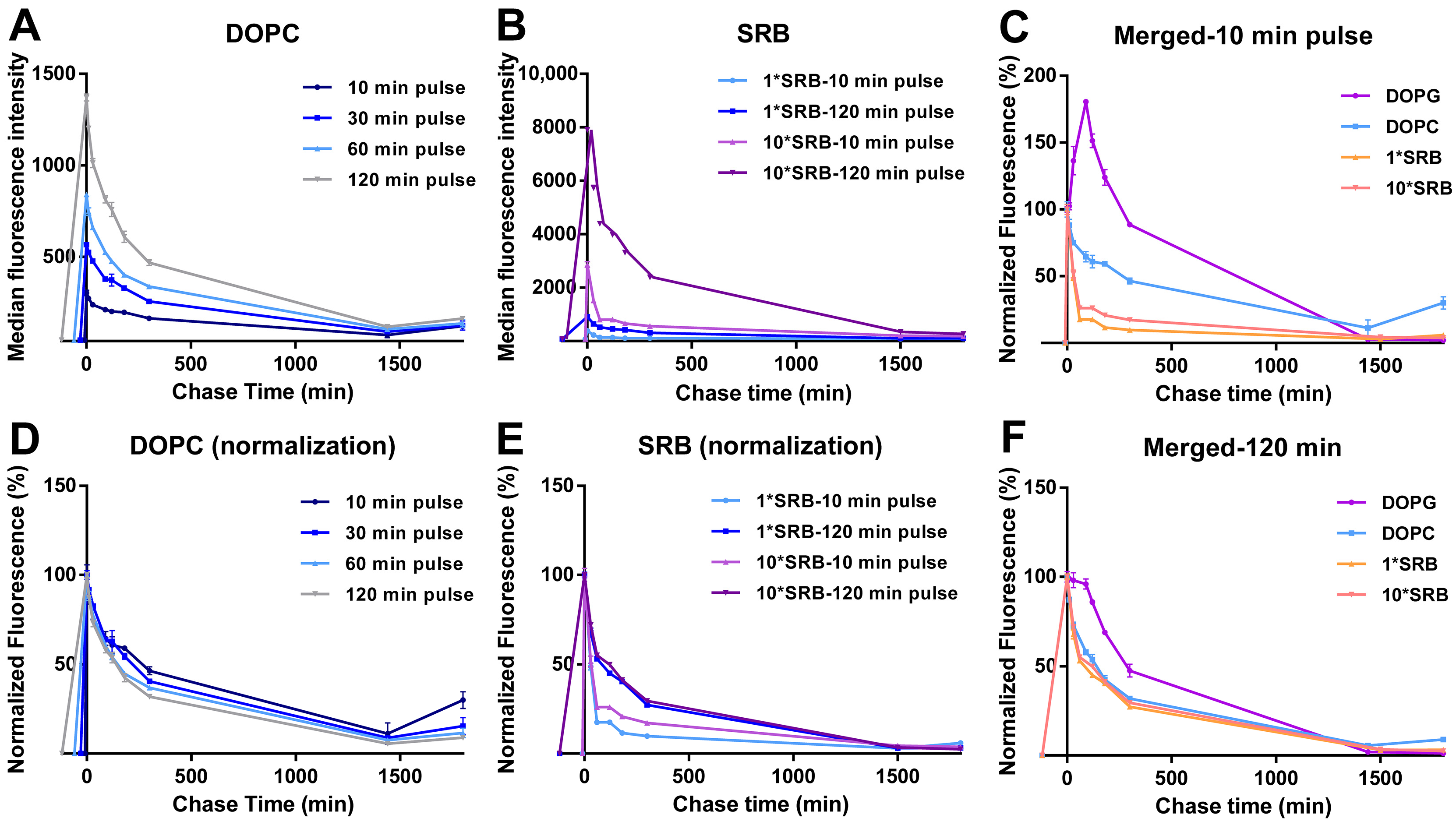
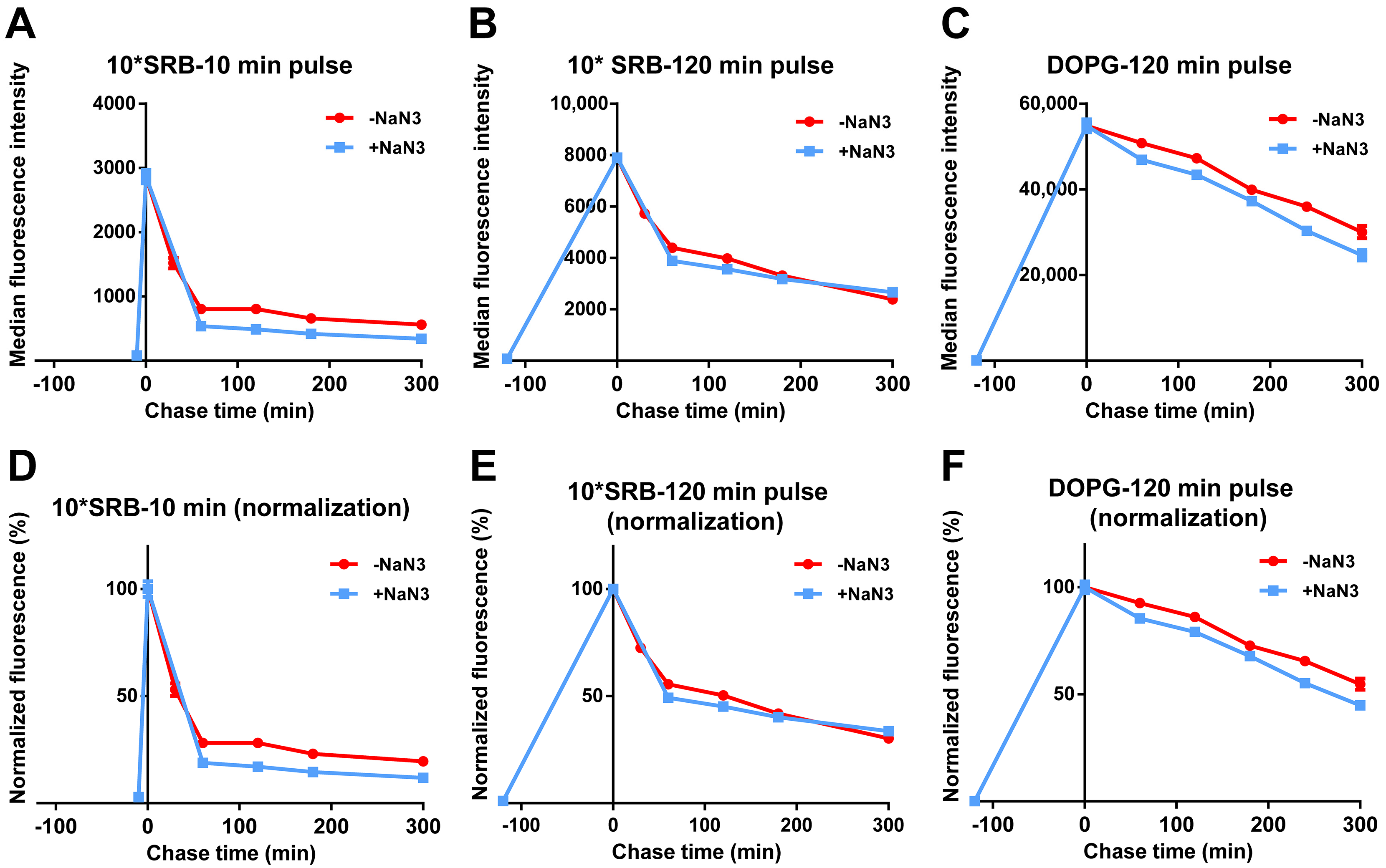
3.2. Intracellular Release of Liposomes
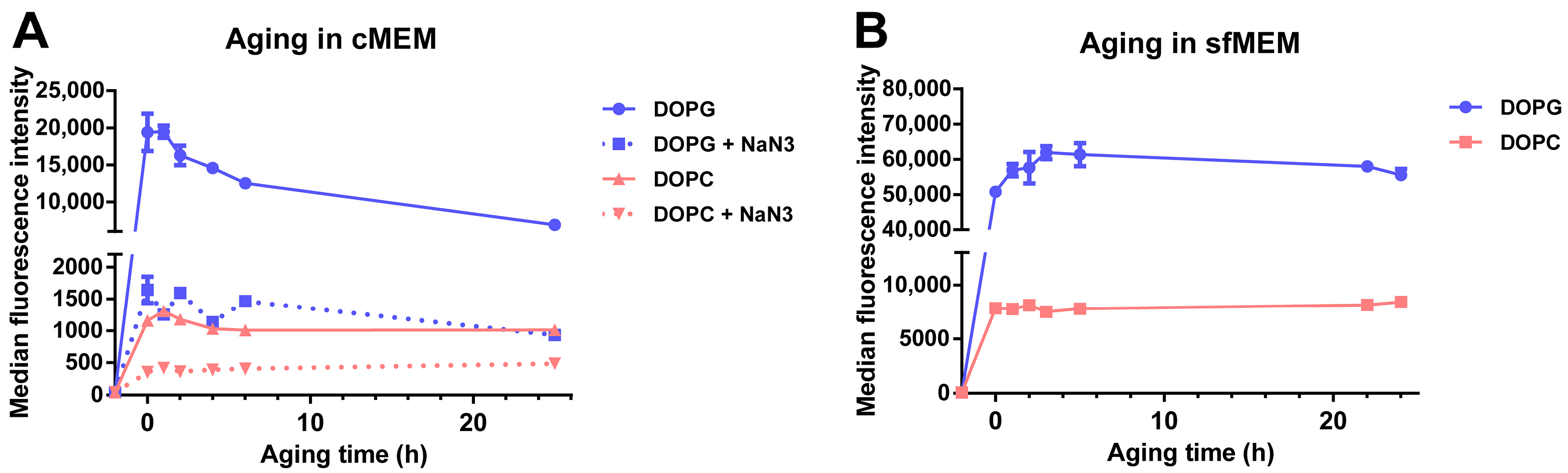
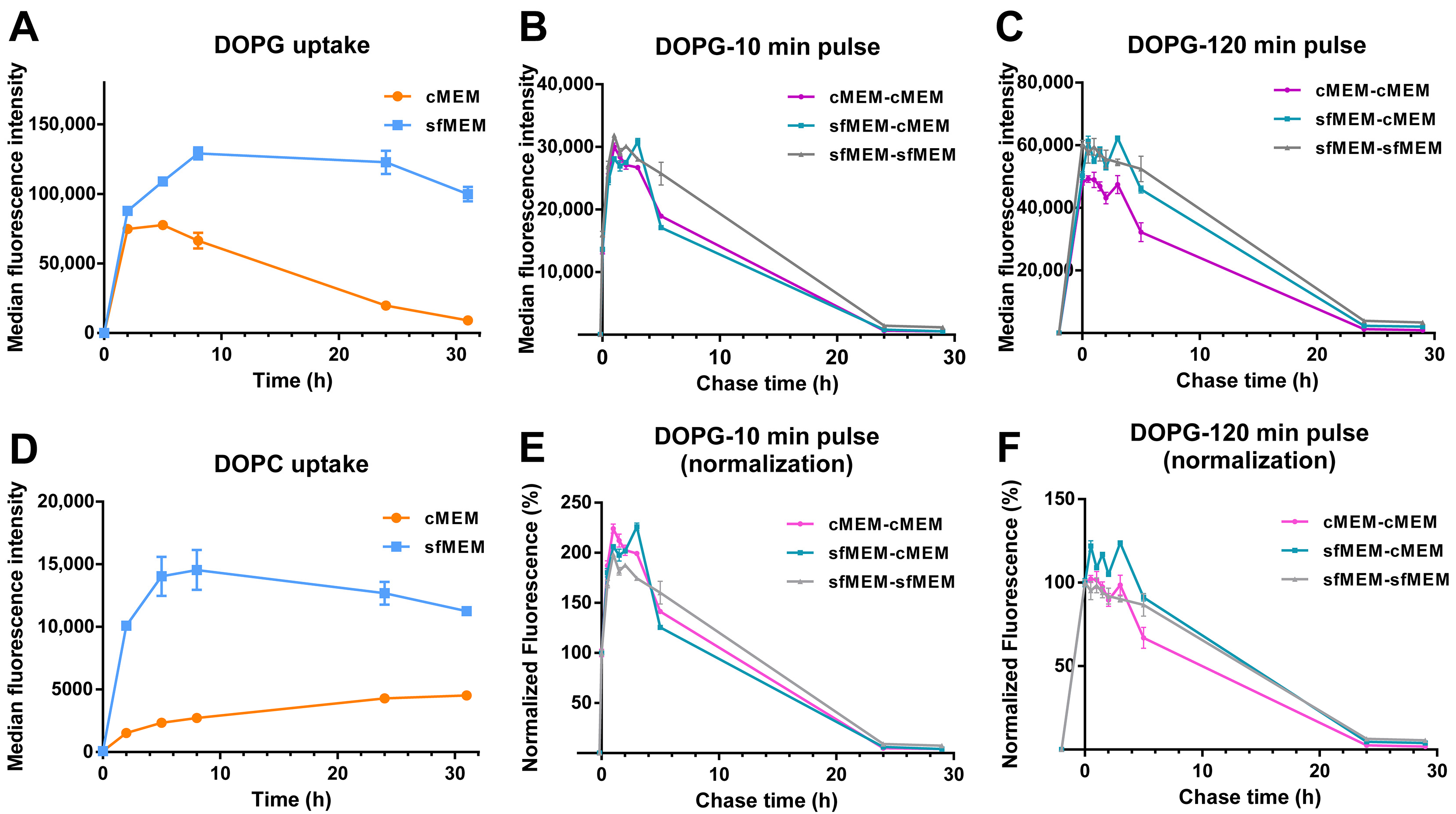
3.3. Stability of Liposomes in Biological Environments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Caster, J.M.; Eblan, M.J.; Wang, A.Z. Clinical Translation of Nanomedicine. Chem. Rev. 2015, 115, 11147–11190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Jamal, W.T.; Kostarelos, K. Liposomes: From a Clinically Established Drug Delivery System to a Nanoparticle Platform for Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G. Liposome-Protein Corona in a Physiological Environment: Challenges and Opportunities for Targeted Delivery of Nanomedicines. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The First FDA-Approved Nano-Drug: Lessons Learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Martin, F.; Huang, A.; Uziely, B.; Kaufman, B.; Safra, T. Prolonged Circulation Time and Enhanced Accumulation in Malignant Exudates of Doxorubicin Encapsulated in Polyethylene-Glycol Coated Liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Torchilin, V.P. Recent Advances with Liposomes as Pharmaceutical Carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Yang, J.; Bahreman, A.; Daudey, G.; Bussmann, J.; Olsthoorn, C.L.; Kros, A. Drug Delivery via Cell Membrane Fusion Using Lipopeptide Modified Liposomes. ACS Central Sci. 2016, 2, 621–630. [Google Scholar] [CrossRef]
- Yang, J.; Shimada, Y.; Olsthoorn, R.C.L.; Snaar-Jagalska, B.E.; Spaink, H.P.; Kros, A. Application of Coiled Coil Peptides in Liposomal Anticancer Drug Delivery Using a Zebrafish Xenograft Model. ACS Nano 2016, 10, 7428–7435. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of Polyplex- and Lipoplex-Mediated Delivery of Nucleic Acids: Real-Time Visualization of Transient Membrane Destabilization without Endosomal Lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef]
- Mo, R.; Jiang, T.; Gu, Z. Enhanced Anticancer Efficacy by ATP-Mediated Liposomal Drug Delivery. Angew. Chem. Int. Ed. 2014, 53, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Wu, Y.; Wang, Y.; Koo, B.; Chen, L.; Petrochenko, P.; Dong, Y.; Choi, S.; Kozak, D.; Oktem, B.; et al. Probing the Mechanism of Bupivacaine Drug Release from Multivesicular Liposomes. J. Control. Release 2019, 294, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.M.; Hultz, M.; Searson, P.C. Leakage Kinetics of the Liposomal Chemotherapeutic Agent Doxil: The Role of Dissolution, Protonation, and Passive Transport, and Implications for Mechanism of Action. J. Control. Release 2018, 269, 171–176. [Google Scholar] [CrossRef]
- Lee, R.J.; Wang, S.; Turk, M.J.; Low, P.S. The Effects of PH and Intraliposomal Buffer Strength on the Rate of Liposome Content Release and Intracellular Drug Delivery. Biosci. Rep. 1998, 18, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Masin, D.; Mcintosh, N.L.; Madden, T.D.; Bally, M.B. Role of Drug Release and Liposome-Mediated Drug Delivery in Governing the Therapeutic Activity of Liposomal Mitoxantrone Used to Treat Human A431 and LS180 Solid Tumors. J. Pharmacol. Exp. Ther. 2000, 292, 337–345. [Google Scholar]
- Kohli, A.G.; Kieler-Ferguson, H.M.; Chan, D.; Szoka, F.C. A Robust and Quantitative Method for Tracking Liposome Contents after Intravenous Administration. J. Control. Release 2014, 176, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Seynhaeve, A.L.B.; Dicheva, B.M.; Hoving, S.; Koning, G.A.; Ten Hagen, T.L.M. Intact Doxil Is Taken up Intracellularly and Released Doxorubicin Sequesters in the Lysosome: Evaluated by in Vitro/in Vivo Live Cell Imaging. J. Control. Release 2013, 172, 330–340. [Google Scholar] [CrossRef]
- Charrois, G.J.R.; Allen, T.M. Drug Release Rate Influences the Pharmacokinetics, Biodistribution, Therapeutic Activity, and Toxicity of Pegylated Liposomal Doxorubicin Formulations in Murine Breast Cancer. Biochim. Biophys. Acta Biomembr. 2004, 1663, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmady, Z.S.; Hadjidemetriou, M.; Gubbins, J.; Kostarelos, K. Formation of Protein Corona in Vivo Affects Drug Release from Temperature-Sensitive Liposomes. J. Control. Release 2018, 276, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Probing the Mechanism of Drug Release from Liposomes. J. Control. Release 2019, 294, 390. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.; Gupta, N.; Mulla, N.S.; Shukla, S.; Guerrero, Y.A.; Gupta, V. Role of In Vitro Release Methods in Liposomal Formulation Development: Challenges and Regulatory Perspective. AAPS J. 2017, 19, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Merodio, M.; Arnedo, A.; Renedo, M.J.; Irache, J.M. Ganciclovir-Loaded Albumin Nanoparticles: Characterization and in Vitro Release Properties. Eur. J. Pharm. Sci. 2001, 12, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hindi, K.M.; Ditto, A.J.; Panzner, M.J.; Medvetz, D.A.; Han, D.S.; Hovis, C.E.; Hilliard, J.K.; Taylor, J.B.; Yun, Y.H.; Cannon, C.L.; et al. The Antimicrobial Efficacy of Sustained Release Silver-Carbene Complex-Loaded l-Tyrosine Polyphosphate Nanoparticles: Characterization, in Vitro and in Vivo Studies. Biomaterials 2009, 30, 3771–3779. [Google Scholar] [CrossRef] [Green Version]
- Washington, C. Evaluation of Non-Sink Dialysis Methods for the Measurement of Drug Release from Colloids: Effects of Drug Partition. Int. J. Pharm. 1989, 56, 71–74. [Google Scholar] [CrossRef]
- Washington, C. Drug Release from Microdisperse Systems: A Critical Review. Int. J. Pharm. 1990, 58, 1–12. [Google Scholar] [CrossRef]
- Moreno-Bautista, G.; Tam, K.C. Evaluation of Dialysis Membrane Process for Quantifying the in Vitro Drug-Release from Colloidal Drug Carriers. Colloids Surf. A Physicochem. Eng. Asp. 2011, 389, 299–303. [Google Scholar] [CrossRef]
- Heeremans, J.L.M.; Gerrttsen, H.R.; Meusen, S.P.; Mijnheer, F.W.; Gangaram Panday, R.S.; Prevost, R.; Kluft, C.; Crommelin, D.J.A. The Preparation of Tissue-Type Plasminogen Activator (t-PA) Containing Liposomes: Entrapment Efficiency and Ultracentrifugation Damage. J. Drug Target. 1995, 3, 301–310. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. A Two-Stage Reverse Dialysis in Vitro Dissolution Testing Method for Passive Targeted Liposomes. Int. J. Pharm. 2012, 426, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Mesquita, B.; Horvatovich, P.; Salvati, A. Tuning Liposome Composition to Modulate Corona Formation in Human Serum and Cellular Uptake. Acta Biomater. 2020, 106, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.C.M. Colorimetric Determination of Phospholipids with Ammonium Ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.A.; Szczepaniak, A.; Króliczewski, J. Comparative Studies on Detergent-Assisted Apocytochrome B6 Reconstitution into Liposomal Bilayers Monitored by Zetasizer Instruments. PLoS ONE 2014, 9, e111341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, M.C.A.; Boekema, E.J. Two Distinct Mechanisms of Vesicle-to-Micelle and Micelle-to-Vesicle Transition Are Mediated by the Packing Parameter of Phospholipid—Detergent Systems. Biochim. Biophys. Acta (BBA)—Biomembr. 2007, 1768, 2681–2689. [Google Scholar] [CrossRef] [Green Version]
- Montizaan, D.; Yang, K.; Reker-Smit, C.; Salvati, A. Comparison of the Uptake Mechanisms of Zwitterionic and Negatively Charged Liposomes by HeLa Cells. Nanomed. Nanotechnol. Biol. Med. 2020, 30, 102300. [Google Scholar] [CrossRef]
- Lesniak, A.; Salvati, A.; Santos-Martinez, M.J.; Radomski, M.W.; Dawson, K.A.; Åberg, C. Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency. J. Am. Chem. Soc. 2013, 135, 1438–1444. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Aberg, C.; Salvati, A.; Dawson, K.A. Role of Cell Cycle on the Cellular Uptake and Dilution of Nanoparticles in a Cell Population. Nat. Nanotechnol. 2012, 7, 62–68. [Google Scholar] [CrossRef]
- Kato, H. In Vitro Assays: Tracking Nanoparticles inside Cells. Nat. Nanotechnol. 2011, 6, 139–140. [Google Scholar] [CrossRef]
- Salvati, A.; Åberg, C.; dos Santos, T.; Varela, J.; Pinto, P.; Lynch, I.; Dawson, K.A. Experimental and Theoretical Comparison of Intracellular Import of Polymeric Nanoparticles and Small Molecules: Toward Models of Uptake Kinetics. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Shapero, K.; Fenaroli, F.; Lynch, I.; Cottell, D.C.; Salvati, A.; Dawson, K.A. Time and Space Resolved Uptake Study of Silica Nanoparticles by Human Cells. Mol. Biosyst. 2011, 7, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding Biophysicochemical Interactions at the Nano–Bio Interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S.; Berggard, T.; Thulin, E.; et al. Understanding the Nanoparticle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cleland, L.G. Serum-Induced Leakage of Liposome Contents. BBA Biomembr. 1980, 597, 418–426. [Google Scholar] [CrossRef]
- Hernfindez-Caselles, T.; Villalafn, J.; Gdmez-Fernfindez, J.C. Influence of Liposome Charge and Composition on Their Interaction with Human Blood Serum Proteins. Mol. Cell Biochem. 1993, 120, 119–126. [Google Scholar] [CrossRef]
- Ruysschaert, T.; Marque, A.; Duteyrat, J.-L.; Lesieur, S.; Winterhalter, M.; Fournier, D. Liposome Retention in Size Exclusion Chromatography. BMC Biotechnol. 2005, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, J.A.; Nozaki, Y.; Tanford, C. Gel-Exclusion Chromatography on S1000 Sephacryl: Application to Phospholipid Vesicles. Anal. Biochem. 1983, 130, 471–474. [Google Scholar] [CrossRef]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Åberg, C.; Dawson, K.A.; Salvati, A. Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef]
- Francia, V.; Yang, K.; Deville, S.; Reker-Smit, C.; Nelissen, I.; Salvati, A. Corona Composition Can Affect the Mechanisms Cells Use to Internalize Nanoparticles. ACS Nano 2019, 13, 11107–11121. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Tran, K.; Salvati, A. Tuning Liposome Stability in Biological Environments and Intracellular Drug Release Kinetics. Biomolecules 2023, 13, 59. https://doi.org/10.3390/biom13010059
Yang K, Tran K, Salvati A. Tuning Liposome Stability in Biological Environments and Intracellular Drug Release Kinetics. Biomolecules. 2023; 13(1):59. https://doi.org/10.3390/biom13010059
Chicago/Turabian StyleYang, Keni, Karolina Tran, and Anna Salvati. 2023. "Tuning Liposome Stability in Biological Environments and Intracellular Drug Release Kinetics" Biomolecules 13, no. 1: 59. https://doi.org/10.3390/biom13010059
APA StyleYang, K., Tran, K., & Salvati, A. (2023). Tuning Liposome Stability in Biological Environments and Intracellular Drug Release Kinetics. Biomolecules, 13(1), 59. https://doi.org/10.3390/biom13010059