
Evaluation of an Adapted Semi-Automated DNA Extraction for Human Salivary Shotgun Metagenomics
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Declaration
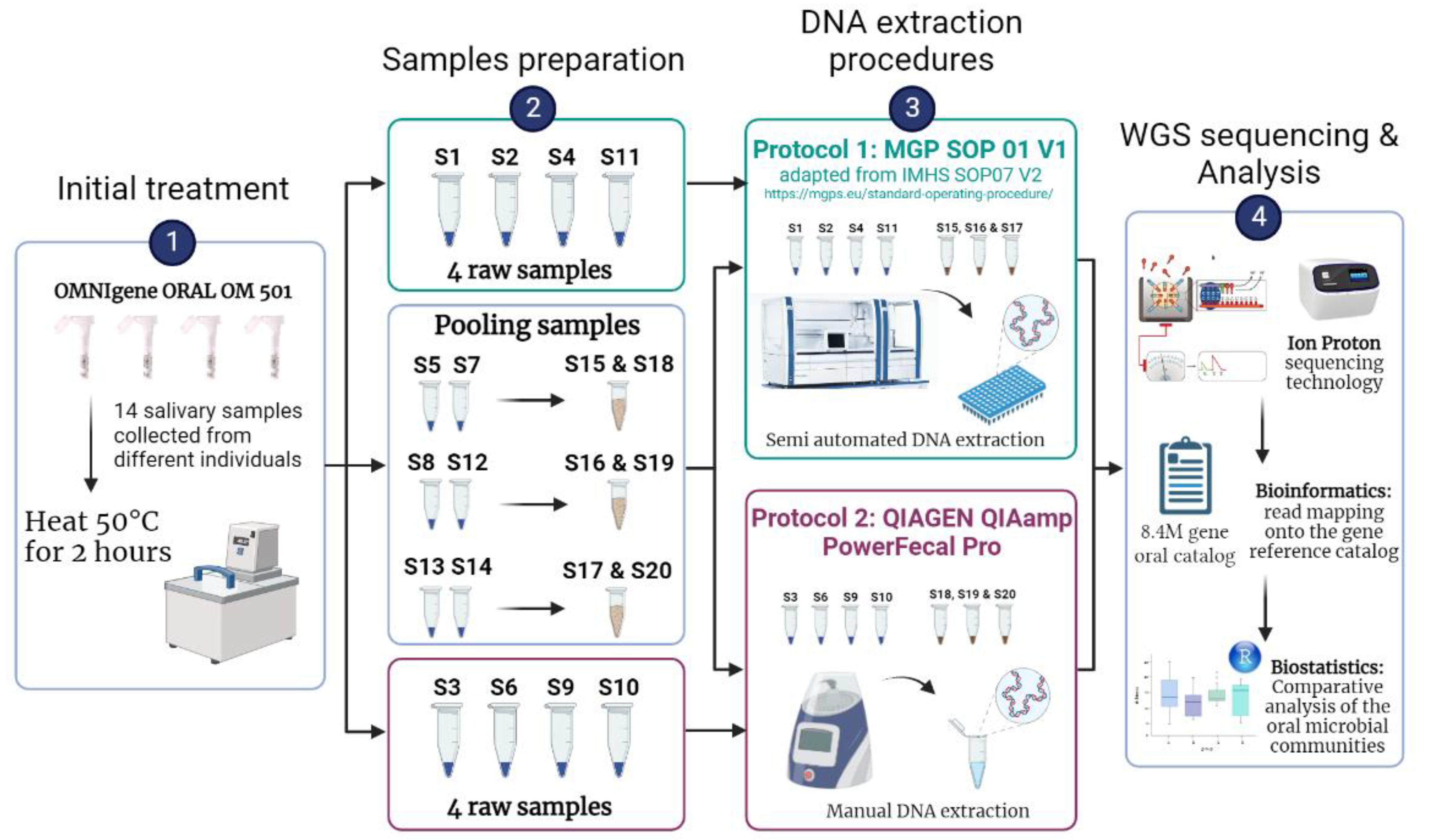
2.2. Salivary Collection Using OMNIgene ORAL OM 501 Device
2.3. Overall Strategy
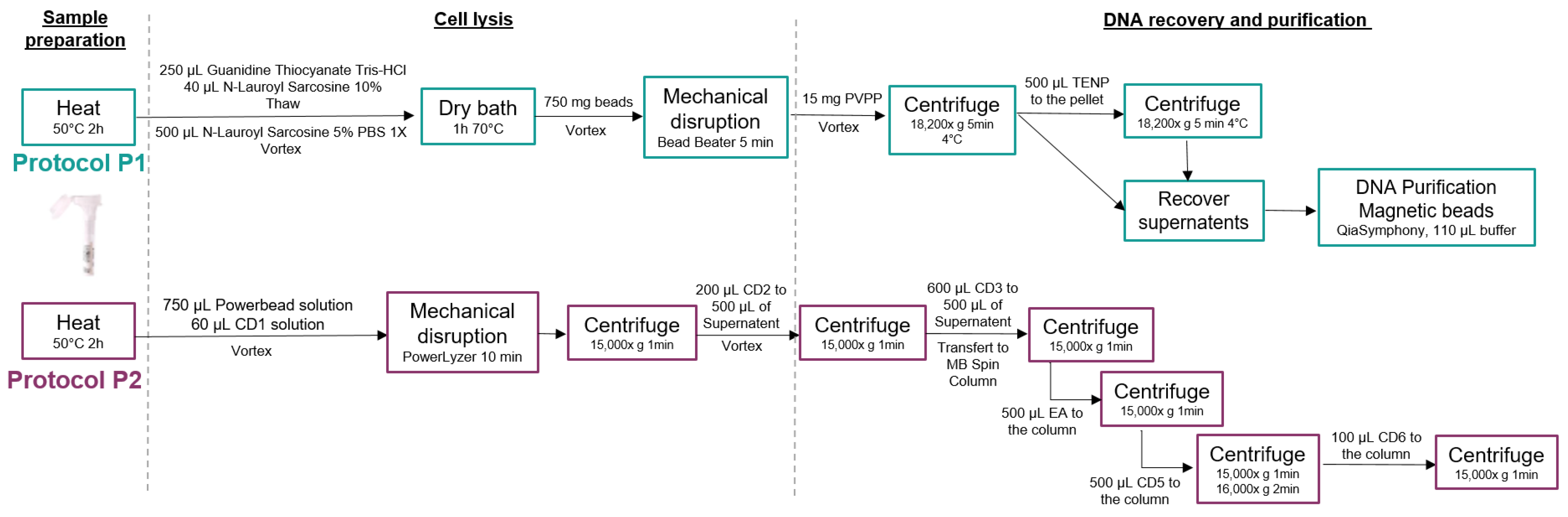
2.4. DNA Extraction Procedures
- Procedure 1: MGP Standard Operating Procedure
- Procedure 2: QIAGEN QIAamp Power Fecal Pro
2.5. DNA Yield and Quality Evaluation
2.6. Shotgun Metagenomics
2.7. Bioinformatics: Read Mapping and Microbial Species
2.8. Biostatistics
3. Results
3.1. Overview of the Analytical Workflow
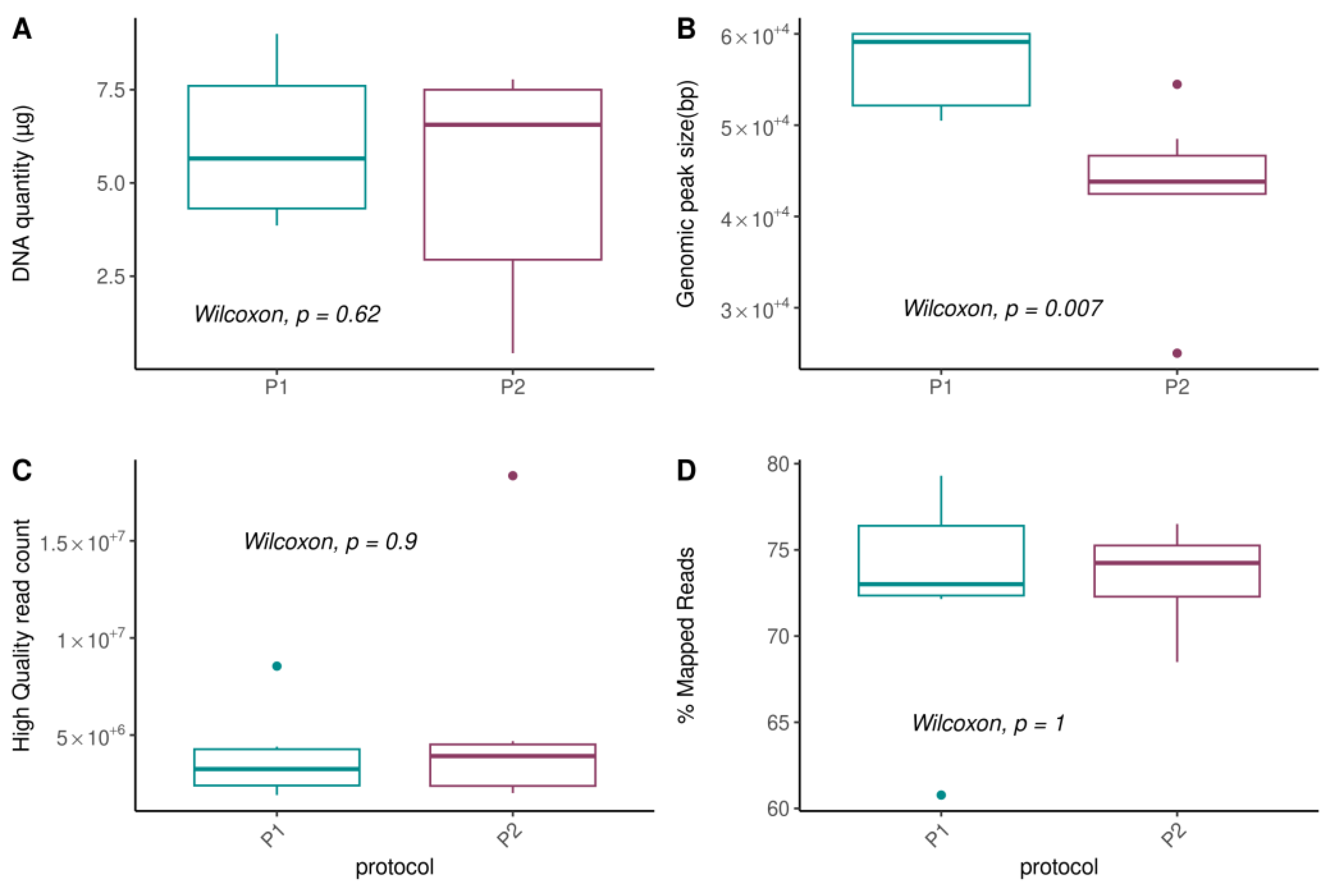
3.2. Evaluation of Standard DNA Extraction Quality Parameters
3.2.1. DNA Concentration and Fragment Size
3.2.2. Metagenomic Read Quality and Mapping
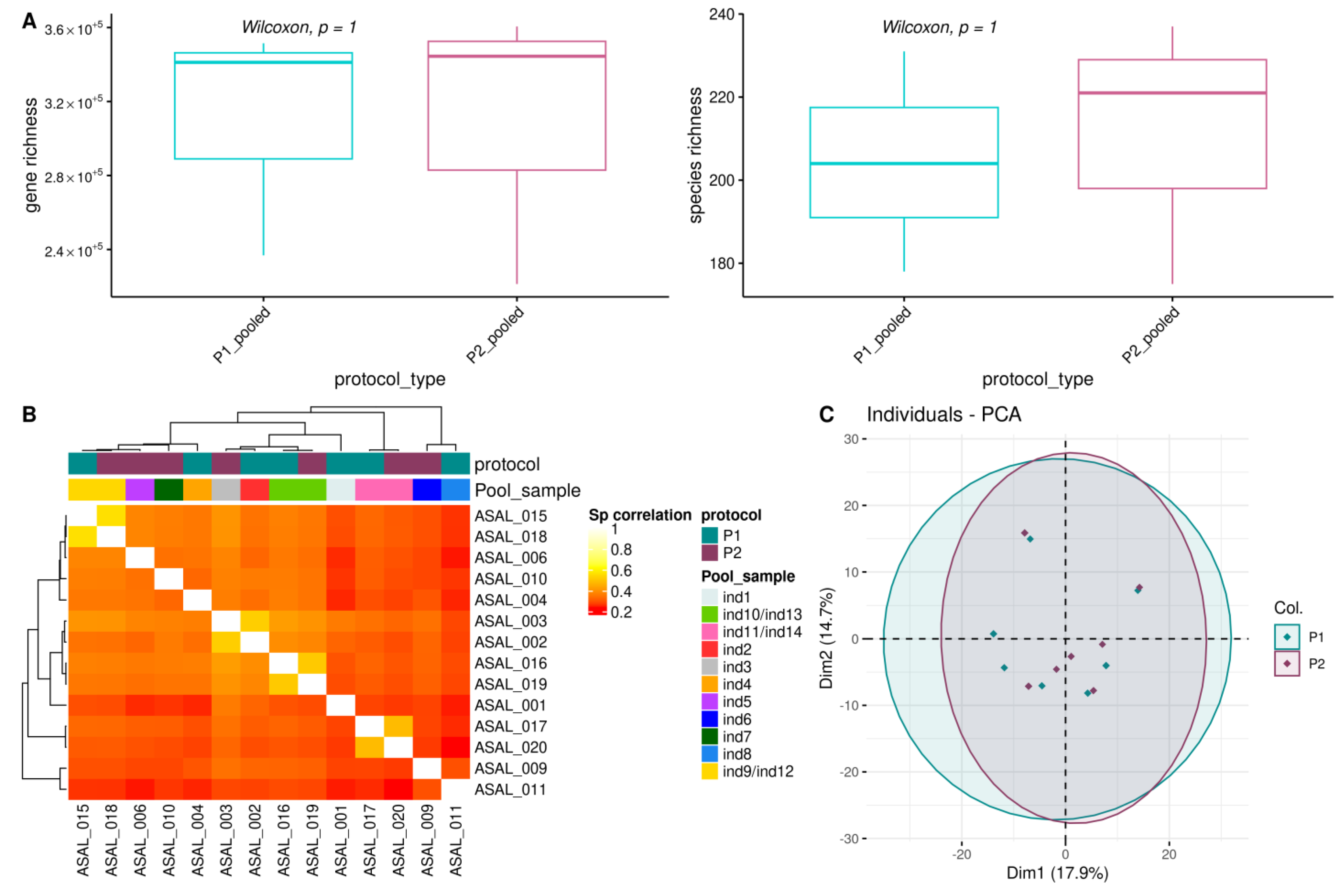
3.3. Impact of DNA Isolation Procedures on the Oral Microbiome
3.3.1. Alpha Diversity Indices
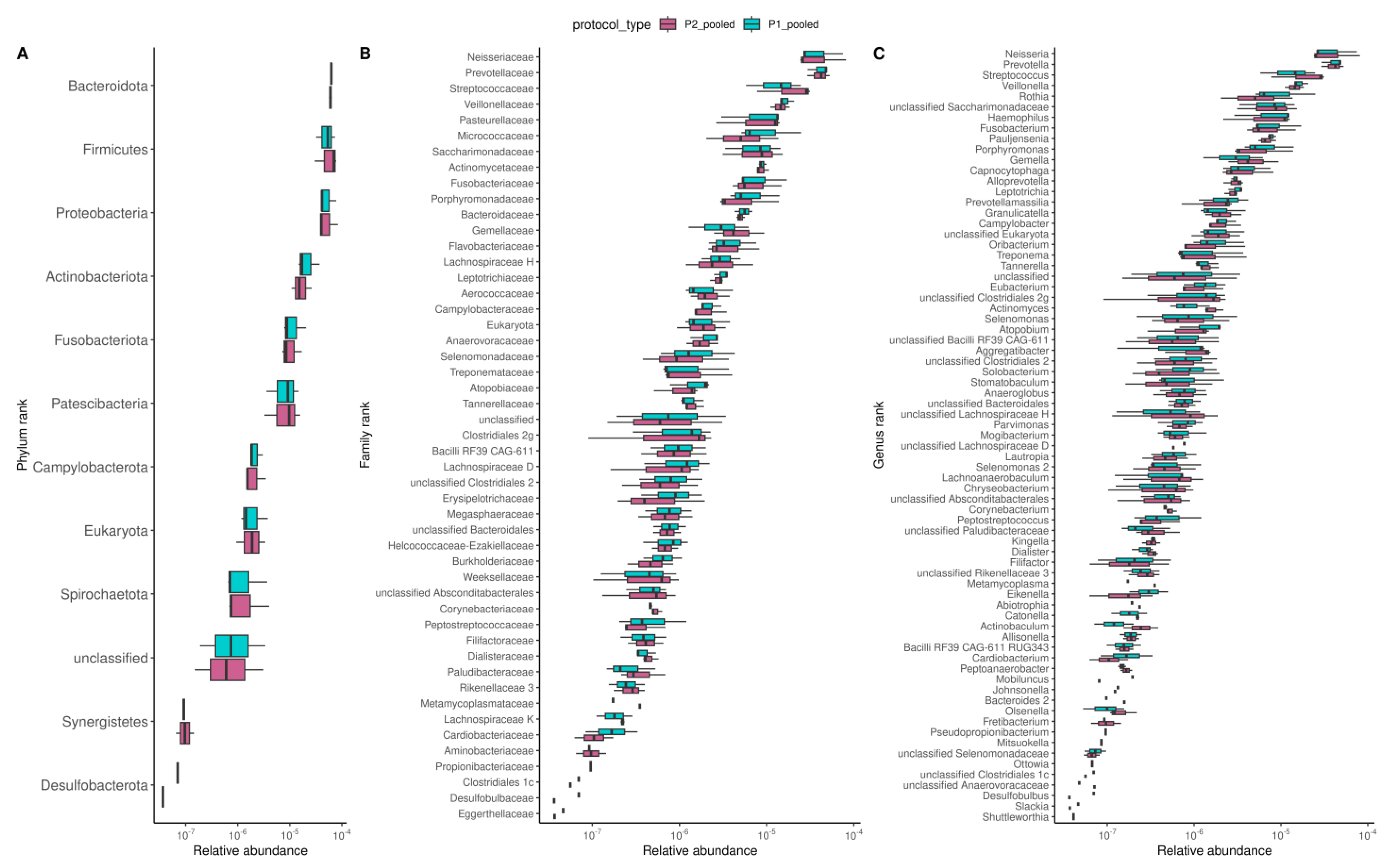
3.3.2. Structure and Composition of the Oral Microbiome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Martu, M.-A.; Maftei, G.-A.; Luchian, I.; Stefanescu, O.M.; Scutariu, M.M.; Solomon, S.M. The Effect of Acknowledged and Novel Anti-Rheumatic Therapies on Periodontal Tissues—A Narrative Review. Pharmaceuticals 2021, 14, 1209. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Sahingur, S.E.; Bajaj, J.S. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight 2017, 2, e94416. [Google Scholar] [CrossRef]
- Michaud, D.S.; Izard, J. Microbiota, oral microbiome, and pancreatic cancer. Cancer J. 2014, 20, 203–206. [Google Scholar] [CrossRef]
- Koliarakis, I.; Messaritakis, I.; Nikolouzakis, T.K.; Hamilos, G.; Souglakos, J.; Tsiaoussis, J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. IJMS 2019, 20, 4146. [Google Scholar] [CrossRef]
- Long, J.; Cai, Q.; Steinwandel, M.; Hargreaves, M.K.; Bordenstein, S.R.; Blot, W.J.; Zheng, W.; Shu, X.O. Association of oral microbiome with type 2 diabetes risk. J. Periodontal Res. 2017, 52, 636–643. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef]
- Mihaila, D.; Donegan, J.; Barns, S.; LaRocca, D.; Du, Q.; Zheng, D.; Vidal, M.; Neville, C.; Uhlig, R.; Middleton, F.A. The oral microbiome of early stage Parkinson’s disease and its relationship with functional measures of motor and non-motor function. PLoS ONE 2019, 14, e0218252. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kang, W.; Hwang, Y.S.; Lee, S.H.; Park, K.W.; Kim, M.S.; Lee, H.; Yoon, H.J.; Park, Y.K.; Chalita, M.; et al. Oral and gut dysbiosis leads to functional alterations in Parkinson’s disease. NPJ Parkinsons Dis. 2022, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 2022, 13, 6958. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, D.K.; Allen, S.J. The Microbiome and Disease: Reviewing the Links between the Oral Microbiome, Aging, and Alzheimer’s Disease. JAD 2014, 43, 725–738. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium, A framework for human microbiome research. Nature 2012, 486, 215–221. [CrossRef] [PubMed]
- Bellagambi, F.G.; Lomonaco, T.; Salvo, P.; Vivaldi, F.; Hangouët, M.; Ghimenti, S.; Biagini, D.; Di Francesco, F.; Fuoco, R.; Errachid, A. Saliva sampling: Methods and devices An overview. TrAC Trends Anal. Chem. 2020, 124, 115781. [Google Scholar] [CrossRef]
- Segata, N.; Haake, S.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome 2012, 13, R42. [Google Scholar] [CrossRef]
- Maki, K.A.; Kazmi, N.; Barb, J.J.; Ames, N. The Oral and Gut Bacterial Microbiomes: Similarities, Differences, and Connections. Biol. Res. Nurs. 2021, 23, 7–20. [Google Scholar] [CrossRef]
- Li, K.; Bihan, M.; Yooseph, S.; Methé, B.A. Analyses of the microbial diversity across the human microbiome. PLoS ONE 2012, 7, e32118. [Google Scholar] [CrossRef]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef]
- Yano, Y.; Hua, X.; Wan, Y.; Suman, S.; Zhu, B.; Dagnall, C.L.; Hutchinson, A.; Jones, K.; Hicks, B.D.; Shi, J.; et al. Comparison of Oral Microbiota Collected Using Multiple Methods and Recommendations for New Epidemiologic Studies. mSystems 2020, 5, e00156-20. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- David, A.; Morabito, C. Protocol for DNA Extraction from Saliva Samples, Used for Shotgun Microbiome Analysis. Protocols.io. 2023. Available online: https://www.protocols.io/view/protocol-for-dna-extraction-from-saliva-samples-us-dm6gpjm11gzp/v1 (accessed on 2 October 2023).
- Higgins, A.L.; Toffoli, M.; Mullin, S.; Lee, C.-Y.; Koletsi, S.; Avenali, M.; Blandini, F.; Schapira, A.H. The remote assessment of parkinsonism supporting the ongoing development of interventions in Gaucher disease. Neurodegener. Dis. Manag. 2021, 11, 451–458. [Google Scholar] [CrossRef]
- Menozzi, E.; Mezabrovschi, R.; Lucas, S.; Macnaughtan, J.; Koletsi, S.; Nazeer, A.; Loefflad, N. Salivary Collection Participant Instructions Using OMNIgene ORAL OM 501 Device. 2023. Available online: https://www.protocols.io/view/salivary-collection-participant-instructions-using-n92ldm8qol5b/v1 (accessed on 2 October 2023).
- Meslier, V.; Quinquis, B.; Da Silva, K.; Oñate, F.P.; Pons, N.; Roume, H.; Podar, M.; Almeida, M. Benchmarking second and third-generation sequencing platforms for microbial metagenomics. Sci. Data 2022, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, A.; Brisse, S. AlienTrimmer: A tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genomics 2013, 102, 500–506. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Almeida, M.; Oñate, F.P.; Pons, N.; Gauthier, F.; Ghozlane, A.; Ehrlich, S.D.; Witherden, E.; Gomez-Cabrero, D. A Catalog of Genes and Species of the Human Oral Microbiota. Data INRAE Recherche Data Gouv. 2021. Available online: https://entrepot.recherche.data.gouv.fr/dataset.xhtml?persistentId=doi:10.15454/WQ4UTV (accessed on 2 October 2023).
- Meslier, V.; Laiola, M.; Roager, H.M.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Plaza Oñate, F.; Le Chatelier, E.; Almeida, M.; Cervino, A.C.L.; Gauthier, F.; Magoulès, F.; Ehrlich, S.D.; Pichaud, M. MSPminer: Abundance-based reconstitution of microbial pan-genomes from shotgun metagenomic data. Bioinformatics 2019, 35, 1544–1552. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- R Core Team. The R Project for Statistical Computing Vienna: R Foundation. 2017. Available online: https://www.r-project.org/ (accessed on 2 October 2022).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.; Solymos, P.; Stevens, M.H.; Wager, H. Vegan: Community Ecology Package; Institute for Statistics and Mathematics: Vienna, Austria, 2019; Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 2 October 2023).
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 107. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 2 October 2023).
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. 2023. Available online: https://cran.r-project.org/package=ggpubr (accessed on 2 October 2023).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-project.org/package=pheatmap (accessed on 2 October 2023).
- Greathouse, K.L.; Sinha, R.; Vogtmann, E. DNA extraction for human microbiome studies: The issue of standardization. Genome Biol. 2019, 20, 212. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.S.; Parmar, V.; Blaser, M.J. Assessing saliva microbiome collection and processing methods. NPJ Biofilms Microbiomes 2021, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, J.; Sato, Y.; Shinozaki, N.; Ye, B.; Tsuboi, A.; Nagasaki, M.; Yamashita, R. Comparison of Boiling and Robotics Automation Method in DNA Extraction for Metagenomic Sequencing of Human Oral Microbes. PLoS ONE 2016, 11, e0154389. [Google Scholar] [CrossRef]
- Lim, Y.; Totsika, M.; Morrison, M.; Punyadeera, C. The saliva microbiome profiles are minimally affected by collection method or DNA extraction protocols. Sci. Rep. 2017, 7, 8523. [Google Scholar] [CrossRef]
- McGaughey, K.D.; Yilmaz-Swenson, T.; Elsayed, N.M.; Cruz, D.A.; Rodriguez, R.R.; Kritzer, M.D.; Peterchev, A.V.; Gray, M.; Lewis, S.R.; Roach, J.; et al. Comparative evaluation of a new magnetic bead-based DNA extraction method from fecal samples for downstream next-generation 16S rRNA gene sequencing. PLoS ONE 2018, 13, e0202858. [Google Scholar] [CrossRef]
- Trigodet, F.; Lolans, K.; Fogarty, E.; Shaiber, A.; Morrison, H.G.; Barreiro, L.; Jabri, B.; Eren, A.M. High molecular weight DNA extraction strategies for long-read sequencing of complex metagenomes. Mol. Ecol. Resour. 2022, 22, 1786–1802. [Google Scholar] [CrossRef]
- Minich, J.J.; Sanders, J.G.; Amir, A.; Humphrey, G.; Gilbert, J.A.; Knight, R. Quantifying and Understanding Well-to-Well Contamination in Microbiome Research. mSystems 2019, 4, e00186-19. [Google Scholar] [CrossRef]
- Godon, J.J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [CrossRef]
- Tegally, H.; San, J.E.; Giandhari, J.; De Oliveira, T. Unlocking the efficiency of genomics laboratories with robotic liquid-handling. BMC Genom. 2020, 21, 729. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tong, X.; Zhu, J.; Tian, L.; Jie, Z.; Zou, Y.; Lin, X.; Liang, H.; Li, W.; Ju, Y.; et al. Metagenome-genome-wide association studies reveal human genetic impact on the oral microbiome. Cell Discov. 2021, 7, 117. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meslier, V.; Menozzi, E.; David, A.; Morabito, C.; Lucas Del Pozo, S.; Famechon, A.; North, J.; Quinquis, B.; Koletsi, S.; Macnaughtan, J.; et al. Evaluation of an Adapted Semi-Automated DNA Extraction for Human Salivary Shotgun Metagenomics. Biomolecules 2023, 13, 1505. https://doi.org/10.3390/biom13101505
Meslier V, Menozzi E, David A, Morabito C, Lucas Del Pozo S, Famechon A, North J, Quinquis B, Koletsi S, Macnaughtan J, et al. Evaluation of an Adapted Semi-Automated DNA Extraction for Human Salivary Shotgun Metagenomics. Biomolecules. 2023; 13(10):1505. https://doi.org/10.3390/biom13101505
Chicago/Turabian StyleMeslier, Victoria, Elisa Menozzi, Aymeric David, Christian Morabito, Sara Lucas Del Pozo, Alexandre Famechon, Janet North, Benoit Quinquis, Sofia Koletsi, Jane Macnaughtan, and et al. 2023. "Evaluation of an Adapted Semi-Automated DNA Extraction for Human Salivary Shotgun Metagenomics" Biomolecules 13, no. 10: 1505. https://doi.org/10.3390/biom13101505
APA StyleMeslier, V., Menozzi, E., David, A., Morabito, C., Lucas Del Pozo, S., Famechon, A., North, J., Quinquis, B., Koletsi, S., Macnaughtan, J., Mezabrovschi, R., Ehrlich, S. D., Schapira, A. H. V., & Almeida, M. (2023). Evaluation of an Adapted Semi-Automated DNA Extraction for Human Salivary Shotgun Metagenomics. Biomolecules, 13(10), 1505. https://doi.org/10.3390/biom13101505