

SF-1 Induces Nuclear PIP2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Materials
2.2. Antibodies
2.3. SF-1 Induction
2.4. Induction of Nuclear PI(4,5)P2 Cross-Reactive Signal by Serum Starvation
2.5. Chamber Slide Fixation and Permeabilization
2.6. Immunofluorescence
2.7. Image Quantitation
2.8. Western Blotting
3. Results
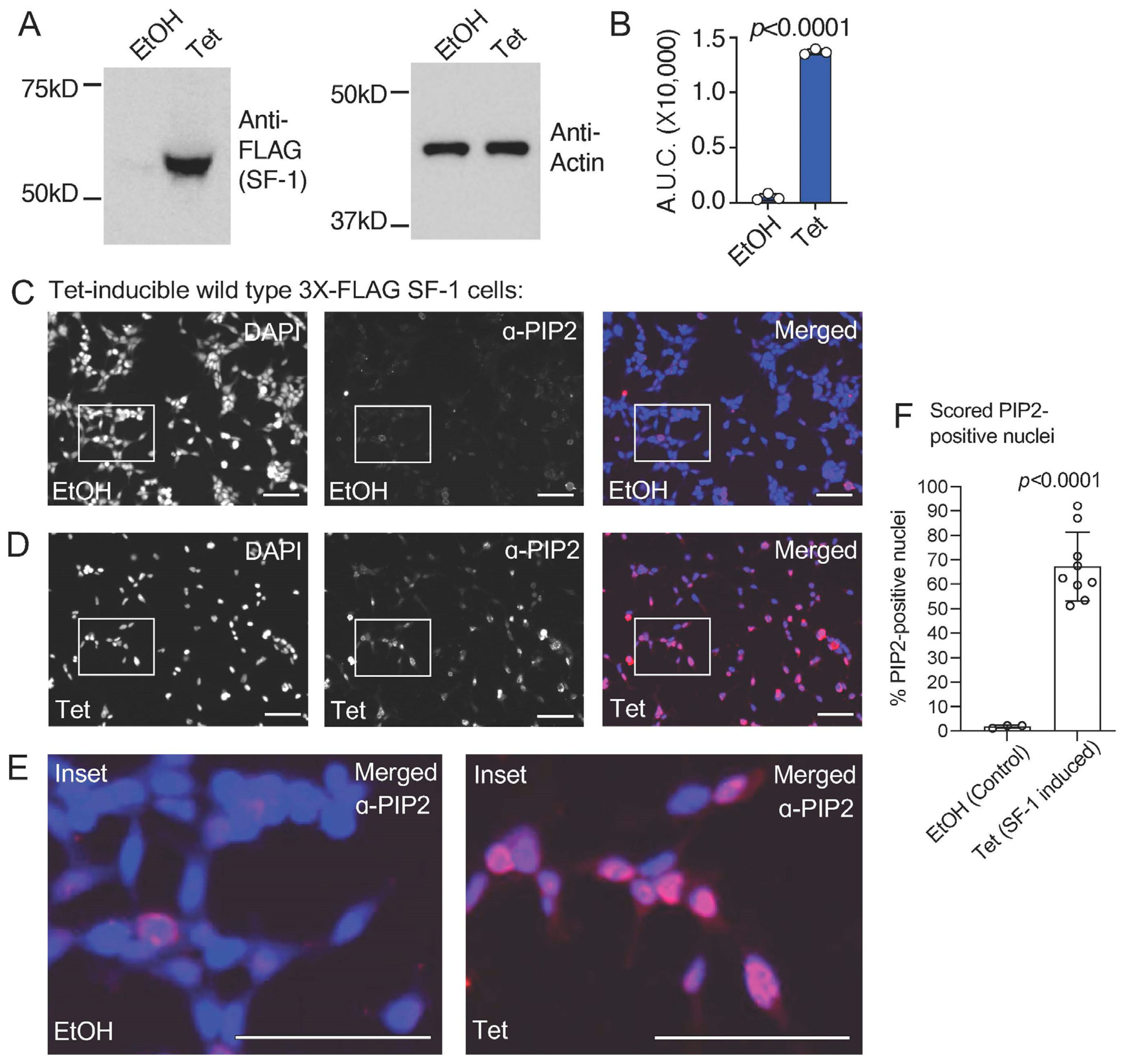
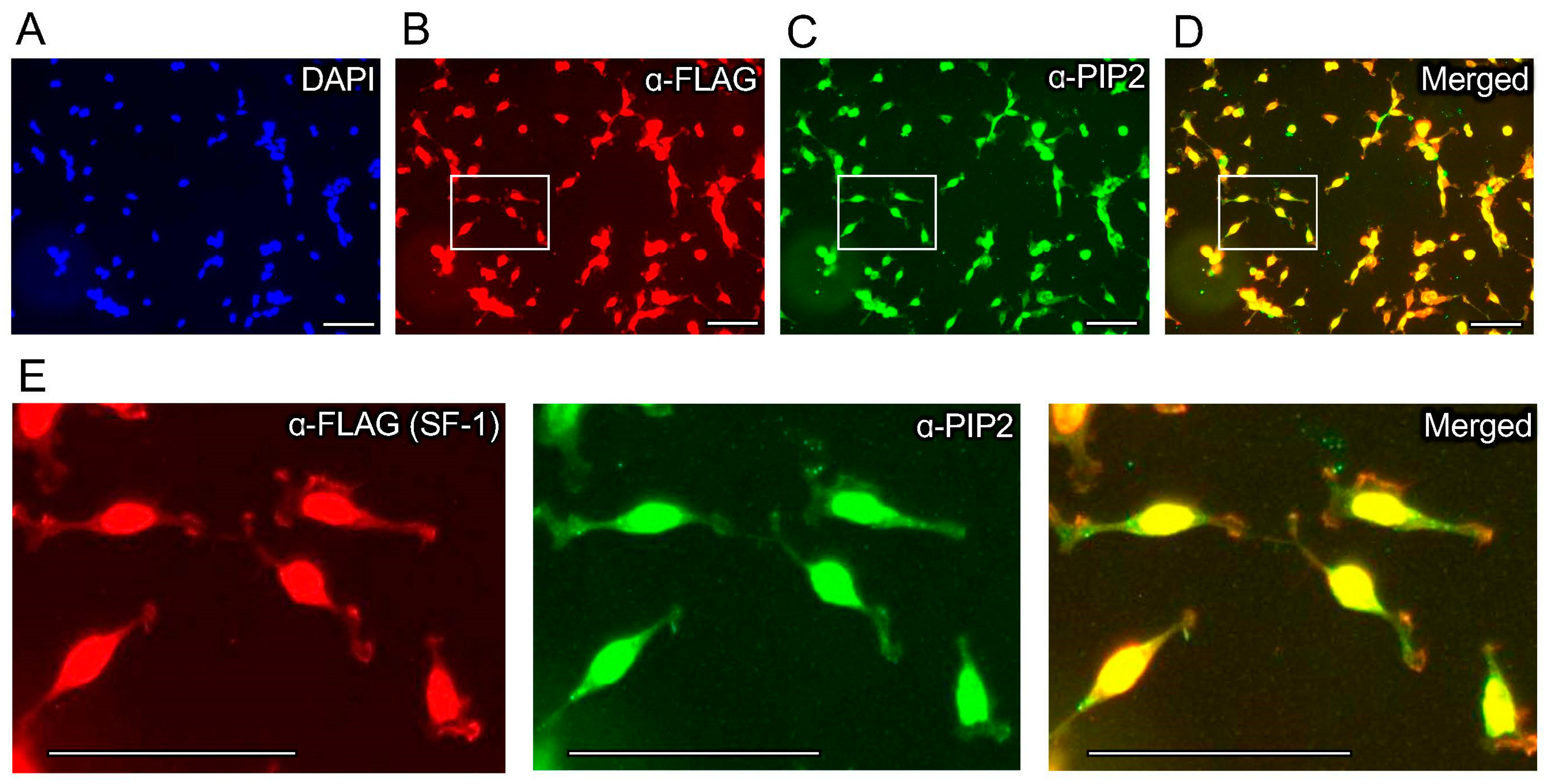
3.1. Ectopic Expression of SF-1 Induces an Immunofluorescence Signal in the Nucleus of HEK293 Cells That Is Cross-Reactive with PI(4,5)P2 Antibodies
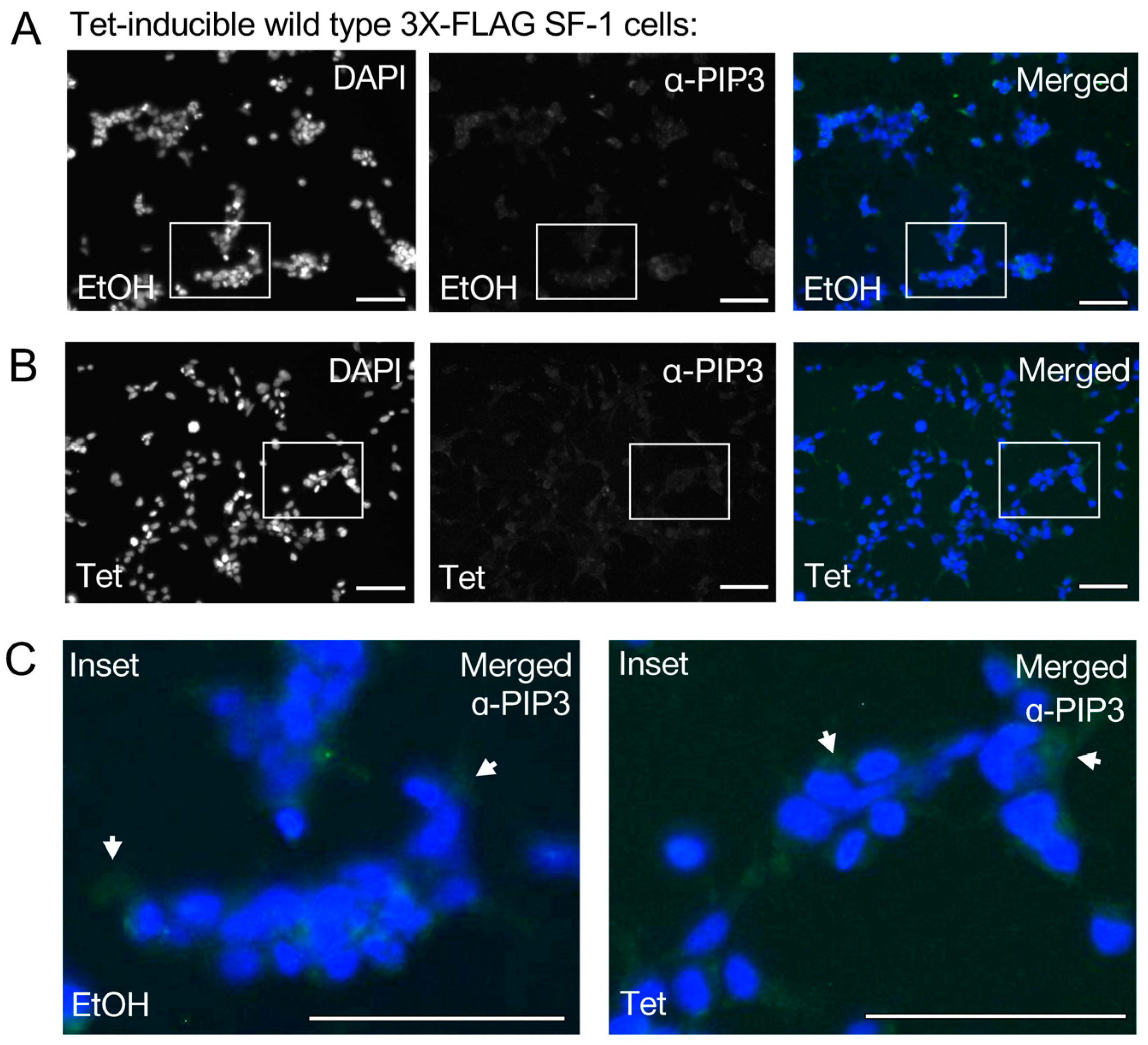
3.2. The Signal Induced by Expression of SF-1 in HEK Cells Does Not Cross-React with PI(3,4,5)P3 Antibodies
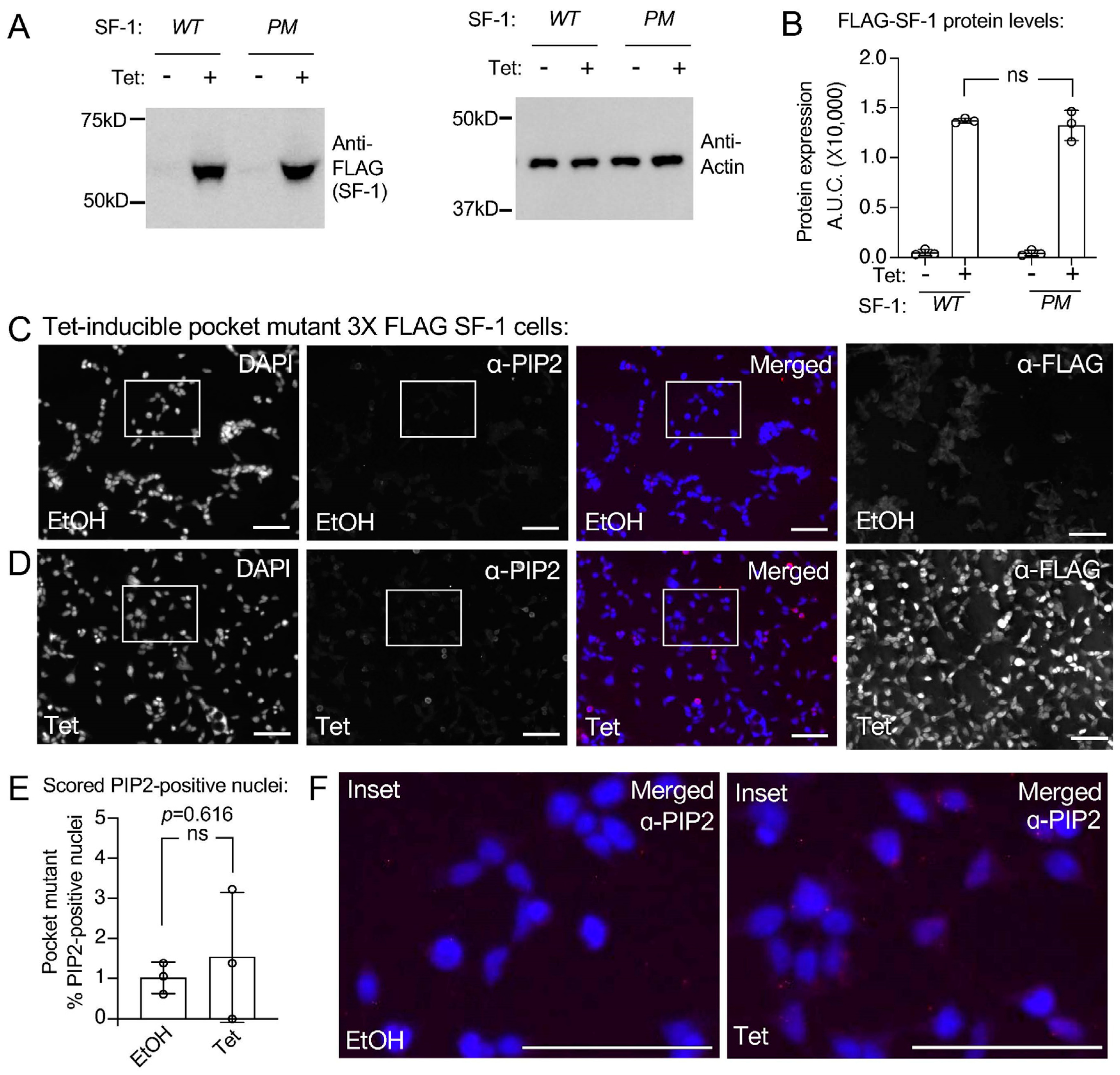
3.3. A Mutant of SF-1 Deficient in Phospholipid Binding Induces Less Signal Cross-Reactive with PI(4,5)P2 Antibodies in HEK Cell Nuclei
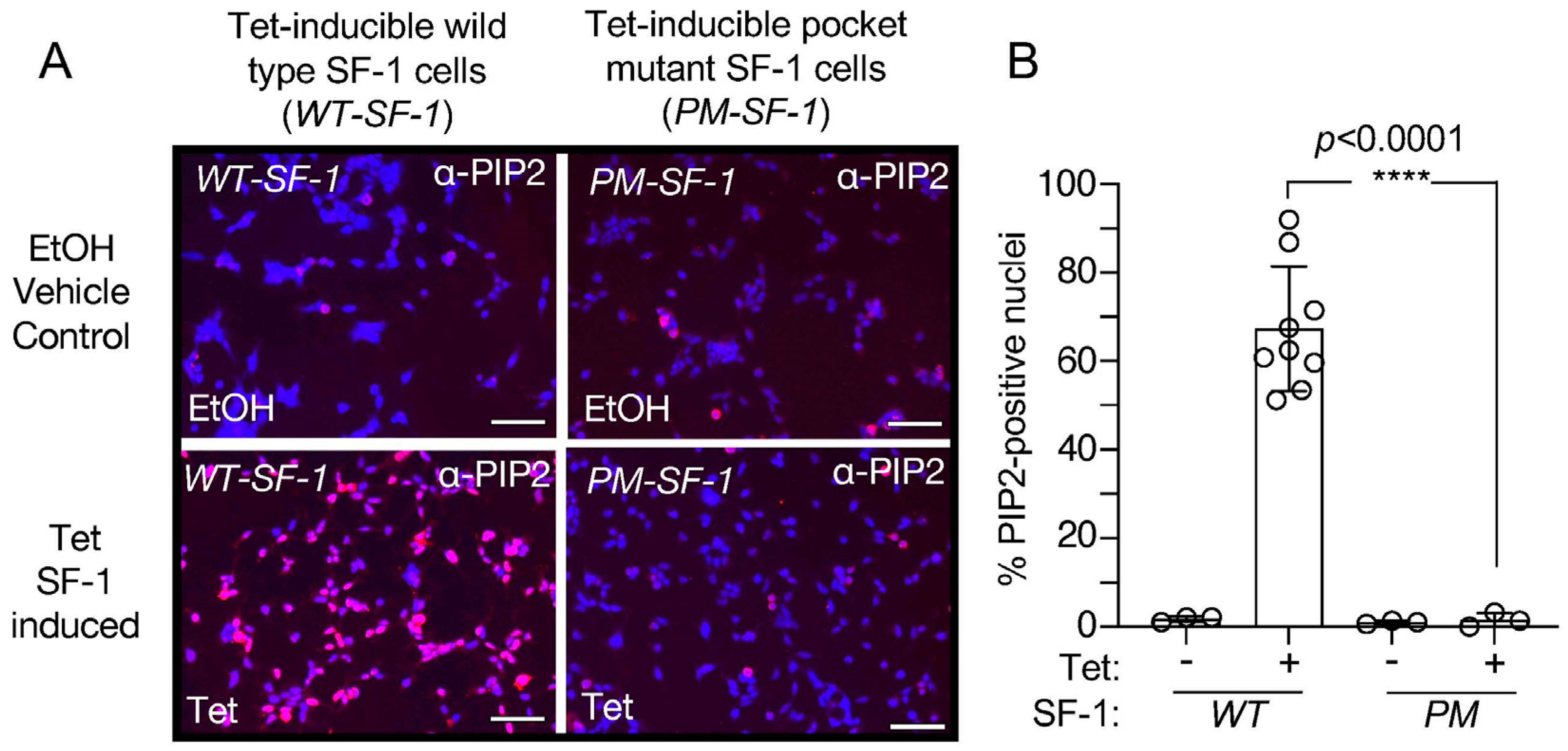
3.4. Direct Comparison of Wild-Type to Pocket Mutant SF-1 Shows Wild-Type SF-1 Induces Significantly More Nuclear PIP2 Antibody Cross-Reactive Signal
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cocco, L.; Gilmour, R.S.; Ognibene, A.; Letcher, A.J.; Manzoli, F.A.; Irvine, R.F. Synthesis of Polyphosphoinositides in Nuclei of Friend Cells. Evidence for Polyphosphoinositide Metabolism inside the Nucleus Which Changes with Cell Differentiation. Biochem. J. 1987, 248, 765–770. [Google Scholar] [CrossRef]
- Lindsay, Y.; McCoull, D.; Davidson, L.; Leslie, N.R.; Fairservice, A.; Gray, A.; Lucocq, J.; Downes, C.P. Localization of Agonist-Sensitive PtdIns(3,4,5)P3 Reveals a Nuclear Pool That Is Insensitive to PTEN Expression. J. Cell Sci. 2006, 119, 5160–5168. [Google Scholar] [CrossRef]
- Vann, L.R.; Wooding, F.B.; Irvine, R.F.; Divecha, N. Metabolism and Compartmentalization of Inositol Lipids in Isolated Rat-Liver Nuclei. Biochem. J. 1997, 327 Pt 2, 569–576. [Google Scholar] [CrossRef]
- Yildirim, S.; Castano, E.; Sobol, M.; Philimonenko, V.V.; Dzijak, R.; Venit, T.; Hozák, P. Involvement of Phosphatidylinositol 4,5-Bisphosphate in RNA Polymerase I Transcription. J. Cell Sci. 2013, 126, 2730–2739. [Google Scholar] [CrossRef]
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide Signaling Pathways in Nuclei Are Associated with Nuclear Speckles. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef]
- Mortier, E.; Wuytens, G.; Leenaerts, I.; Hannes, F.; Heung, M.Y.; Degeest, G.; David, G.; Zimmermann, P. Nuclear Speckles and Nucleoli Targeting by PIP2–PDZ Domain Interactions. EMBO J. 2005, 24, 2556–2565. [Google Scholar] [CrossRef]
- Toska, E.; Campbell, H.A.; Shandilya, J.; Goodfellow, S.J.; Shore, P.; Medler, K.F.; Roberts, S.G. Repression of Transcription by WT1-BASP1 Requires the Myristoylation of BASP1 and the PIP2-Dependent Recruitment of Histone Deacetylase. Cell Rep. 2012, 2, 462–469. [Google Scholar] [CrossRef]
- Rando, O.J.; Zhao, K.; Janmey, P.; Crabtree, G.R. Phosphatidylinositol-Dependent Actin Filament Binding by the SWI/SNF-like BAF Chromatin Remodeling Complex. Proc. Natl. Acad. Sci. USA 2002, 99, 2824–2829. [Google Scholar] [CrossRef]
- Gozani, O.; Karuman, P.; Jones, D.R.; Ivanov, D.; Cha, J.; Lugovskoy, A.A.; Baird, C.L.; Zhu, H.; Field, S.J.; Lessnick, S.L.; et al. The PHD Finger of the Chromatin-Associated Protein ING2 Functions as a Nuclear Phosphoinositide Receptor. Cell 2003, 114, 99–111. [Google Scholar] [CrossRef]
- Vidalle, M.C.; Sheth, B.; Fazio, A.; Marvi, M.V.; Leto, S.; Koufi, F.-D.; Neri, I.; Casalin, I.; Ramazzotti, G.; Follo, M.Y.; et al. Nuclear Phosphoinositides as Key Determinants of Nuclear Functions. Biomolecules 2023, 13, 1049. [Google Scholar] [CrossRef]
- Balaban, C.; Sztacho, M.; Blažíková, M.; Hozák, P. The F-Actin-Binding MPRIP Forms Phase-Separated Condensates and Associates with PI(4,5)P2 and Active RNA Polymerase II in the Cell Nucleus. Cells 2021, 10, 848. [Google Scholar] [CrossRef] [PubMed]
- Stijf-Bultsma, Y.; Sommer, L.; Tauber, M.; Baalbaki, M.; Giardoglou, P.; Jones, D.R.; Gelato, K.A.; van Pelt, J.; Shah, Z.; Rahnamoun, H.; et al. The Basal Transcription Complex Component TAF3 Transduces Changes in Nuclear Phosphoinositides into Transcriptional Output. Mol. Cell 2015, 58, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-Y.; Liu, X.; Cheng, D.; Peng, J.; Chan, P.-K.; Wade, P.A.; Ye, K. Nucleophosmin/B23, a Nuclear PI(3,4,5)P(3) Receptor, Mediates the Antiapoptotic Actions of NGF by Inhibiting CAD. Mol. Cell 2005, 18, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Mellman, D.L.; Gonzales, M.L.; Song, C.; Barlow, C.A.; Wang, P.; Kendziorski, C.; Anderson, R.A. A PtdIns4,5P2-Regulated Nuclear Poly(A) Polymerase Controls Expression of Select MRNAs. Nature 2008, 451, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, W.; Rando, O.J.; Xue, Y.; Swiderek, K.; Kuo, A.; Crabtree, G.R. Rapid and Phosphoinositol-Dependent Binding of the SWI/SNF-like BAF Complex to Chromatin after T Lymphocyte Receptor Signaling. Cell 1998, 95, 625–636. [Google Scholar] [CrossRef]
- Blind, R.D.; Sablin, E.P.; Kuchenbecker, K.M.; Chiu, H.J.; Deacon, A.M.; Das, D.; Fletterick, R.J.; Ingraham, H.A. The Signaling Phospholipid PIP3 Creates a New Interaction Surface on the Nuclear Receptor NR5A1 (SF-1). Proc. Natl. Acad. Sci. USA 2014, 111, 15054–15059. [Google Scholar] [CrossRef]
- Sztacho, M.; Sobol, M.; Balaban, C.; Escudeiro Lopes, S.E.; Hozák, P. Nuclear Phosphoinositides and Phase Separation: Important Players in Nuclear Compartmentalization. Adv. Biol. Regul. 2019, 71, 111–117. [Google Scholar] [CrossRef]
- Krylova, I.N.; Sablin, E.P.; Moore, J.; Xu, R.X.; Waitt, G.M.; MacKay, J.A.; Juzumiene, D.; Bynum, J.M.; Madauss, K.; Montana, V.; et al. Structural Analyses Reveal Phosphatidyl Inositols as Ligands for the NR5A Orphan Receptors SF-1 and LRH-1. Cell 2005, 120, 343–355. [Google Scholar] [CrossRef]
- Sablin, E.P.; Blind, R.D.; Krylova, I.N.; Ingraham, J.G.; Cai, F.; Williams, J.D.; Fletterick, R.J.; Ingraham, H.A. Structure of SF-1 Bound by Different Phospholipids: Evidence for Regulatory Ligands. Mol. Endocrinol. 2009, 23, 25–34. [Google Scholar] [CrossRef]
- Ikeda, Y. NR5A1 (SF-1): A Key Regulator of Development and Function in the Mammalian Reproductive System. Acta Paediatr. Jpn. 1996, 38, 412–419. [Google Scholar] [CrossRef]
- Blind, R.D.D.; Pineda-Torra, I.; Xu, Y.; Xu, H.E.E.; Garabedian, M.J.J. Ligand Structural Motifs Can Decouple Glucocorticoid Receptor Transcriptional Activation from Target Promoter Occupancy. Biochem. Biophys. Res. Commun. 2012, 420, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M. The Nuclear Receptor Superfamily: A Rosetta Stone for Physiology. Mol. Endocrinol. 2005, 19, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- Fujikawa, T.; Castorena, C.M.; Pearson, M.; Kusminski, C.M.; Ahmed, N.; Battiprolu, P.K.; Kim, K.W.; Lee, S.; Hill, J.A.; Scherer, P.E.; et al. SF-1 Expression in the Hypothalamus Is Required for Beneficial Metabolic Effects of Exercise. eLife 2016, 5, e18206. [Google Scholar] [CrossRef] [PubMed]
- Sadovsky, Y.; Crawford, P.A.; Woodson, K.G.; Polish, J.A.; Clements, M.A.; Tourtellotte, L.M.; Simburger, K.; Milbrandt, J. Mice Deficient in the Orphan Receptor Steroidogenic Factor 1 Lack Adrenal Glands and Gonads but Express P450 Side-Chain-Cleavage Enzyme in the Placenta and Have Normal Embryonic Serum Levels of Corticosteroids. Proc. Natl. Acad. Sci. USA 1995, 92, 10939–10943. [Google Scholar] [CrossRef]
- Ferraz-de-Souza, B.; Lin, L.; Achermann, J.C. Steroidogenic Factor-1 (SF-1, NR5A1) and Human Disease. Mol. Cell. Endocrinol. 2011, 336, 198–205. [Google Scholar] [CrossRef]
- Majdic, G.; Young, M.; Gomez-Sanchez, E.; Anderson, P.; Szczepaniak, L.S.; Dobbins, R.L.; McGarry, J.D.; Parker, K.L. Knockout Mice Lacking Steroidogenic Factor 1 Are a Novel Genetic Model of Hypothalamic Obesity. Endocrinology 2002, 143, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ikeda, Y.; Schlosser, D.A.; Parker, K.L. Steroidogenic Factor 1 Is the Essential Transcript of the Mouse Ftz-F1 Gene. Mol. Endocrinol. 1995, 9, 1233–1239. [Google Scholar] [CrossRef]
- Kim, K.W.; Zhao, L.; Parker, K.L. Central Nervous System-Specific Knockout of Steroidogenic Factor 1. Mol. Cell. Endocrinol. 2009, 300, 132–136. [Google Scholar] [CrossRef]
- Kim, K.W.; Li, S.; Zhao, H.; Peng, B.; Tobet, S.A.; Elmquist, J.K.; Parker, K.L.; Zhao, L. CNS-Specific Ablation of Steroidogenic Factor 1 Results in Impaired Female Reproductive Function. Mol. Endocrinol. 2010, 24, 1240–1250. [Google Scholar] [CrossRef]
- Kim, K.W.; Zhao, L.; Donato, J.; Kohno, D.; Xu, Y.; Elias, C.F.; Lee, C.; Parker, K.L.; Elmquist, J.K. Steroidogenic Factor 1 Directs Programs Regulating Diet-Induced Thermogenesis and Leptin Action in the Ventral Medial Hypothalamic Nucleus. Proc. Natl. Acad. Sci. USA 2011, 108, 10673–10678. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shen, W.H.; Ingraham, H.A.; Parker, K.L. Developmental Expression of Mouse Steroidogenic Factor-1, an Essential Regulator of the Steroid Hydroxylases. Mol. Endocrinol. 1994, 8, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Hanley, N.A.; Ikeda, Y.; Luo, X.; Parker, K.L. Steroidogenic Factor 1 (SF-1) Is Essential for Ovarian Development and Function. Mol. Cell. Endocrinol. 2000, 163, 27–32. [Google Scholar] [CrossRef]
- Luo, X.; Ikeda, Y.; Parker, K.L. A Cell-Specific Nuclear Receptor Is Essential for Adrenal and Gonadal Development and Sexual Differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Duregon, E.; Volante, M.; Giorcelli, J.; Terzolo, M.; Lalli, E.; Papotti, M. Diagnostic and Prognostic Role of Steroidogenic Factor 1 in Adrenocortical Carcinoma: A Validation Study Focusing on Clinical and Pathologic Correlates. Hum. Pathol. 2013, 44, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Doghman, M.; Cazareth, J.; Douguet, D.; Madoux, F.; Hodder, P.; Lalli, E. Inhibition of Adrenocortical Carcinoma Cell Proliferation by Steroidogenic Factor-1 NR5A1 (SF-1) Inverse Agonists. J. Clin. Endocrinol. Metab. 2009, 94, 2178–2183. [Google Scholar] [CrossRef]
- Bryant, J.M.; Blind, R.D. Signaling through Non-Membrane Nuclear Phosphoinositide Binding Proteins in Human Health and Disease. J. Lipid Res. 2019, 60, 299–311. [Google Scholar] [CrossRef]
- Bulun, S.E.; Utsunomiya, H.; Lin, Z.; Yin, P.; Cheng, Y.-H.; Pavone, M.E.; Tokunaga, H.; Trukhacheva, E.; Attar, E.; Gurates, B.; et al. Steroidogenic Factor-1 and Endometriosis. Mol. Cell. Endocrinol. 2009, 300, 104–108. [Google Scholar] [CrossRef]
- Lin, B.C.C.; Suzawa, M.; Blind, R.D.D.; Tobias, S.C.C.; Bulun, S.E.E.; Scanlan, T.S.S.; Ingraham, H.A.A. Stimulating the GPR30 Estrogen Receptor with a Novel Tamoxifen Analogue Activates NR5A and Promotes Endometrial Cell Proliferation. Cancer Res. 2009, 69, 5415–5423. [Google Scholar] [CrossRef]
- Lala, D.S.; Rice, D.A.; Parker, K.L. Steroidogenic Factor i, a Key Regulator of Steroidogenic Enzyme Expression, Is the Mouse Homolog of Fushi Tarazu-Factor i. Mol. Endocrinol. 1992, 6, 1249–1258. [Google Scholar] [CrossRef]
- Seacrist, C.D.; Kuenze, G.; Hoffmann, R.M.; Moeller, B.E.; Burke, J.E.; Meiler, J.; Blind, R.D. Integrated Structural Modeling of Full-Length LRH-1 Reveals Inter-Domain Interactions Contribute to Receptor Structure and Function. Structure 2020, 28, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.M.; Malabanan, M.M.; Vanderloop, B.H.; Nichols, C.M.; Haratipour, Z.; Poon, K.T.; Sherrod, S.D.; McLean, J.A.; Blind, R.D. The Acyl Chains of Phosphoinositide PIP3 Alter the Structure and Function of Nuclear Receptor Steroidogenic Factor-1 (NR5A1). J. Lipid Res. 2021, 62, 100081. [Google Scholar] [CrossRef] [PubMed]
- Blind, R.D.; Suzawa, M.; Ingraham, H.A. Direct Modification and Activation of a Nuclear Receptor-PIP2 Complex by the Inositol Lipid Kinase IPMK. Sci. Signal 2012, 5, ra44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Hariharan, A.; Bastianello, G.; Toyama, Y.; Shivashankar, G.V.; Foiani, M.; Sheetz, M.P. DNA Damage Causes Rapid Accumulation of Phosphoinositides for ATR Signaling. Nat. Commun. 2017, 8, 2118. [Google Scholar] [CrossRef] [PubMed]
- Urs, A.N.; Dammer, E.; Kelly, S.; Wang, E.; Merrill, A.H.; Sewer, M.B. Steroidogenic Factor-1 Is a Sphingolipid Binding Protein. Mol. Cell. Endocrinol. 2007, 265–266, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Urs, A.N.; Dammer, E.; Sewer, M.B. Sphingosine Regulates the Transcription of CYP17 by Binding to Steroidogenic Factor-1. Endocrinology 2006, 147, 5249–5258. [Google Scholar] [CrossRef]
- Barneda, D.; Cosulich, S.; Stephens, L.; Hawkins, P. How Is the Acyl Chain Composition of Phosphoinositides Created and Does It Matter? Biochem. Soc. Trans. 2019, 47, 1291–1305. [Google Scholar] [CrossRef]
- Kielkowska, A.; Niewczas, I.; Anderson, K.E.; Durrant, T.N.; Clark, J.; Stephens, L.R.; Hawkins, P.T. A New Approach to Measuring Phosphoinositides in Cells by Mass Spectrometry. Adv. Biol. Regul. 2014, 54, 131–141. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s Tyrosine Kinase (Btk) and Activation by Soluble Inositol Hexakisphosphate. eLife 2015, 4, e06074. [Google Scholar] [CrossRef]
- Lucki, N.C.; Sewer, M.B. Nuclear Sphingolipid Metabolism. Annu. Rev. Physiol. 2012, 74, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Sablin, E.P.; Blind, R.D.; Uthayaruban, R.; Chiu, H.-J.; Deacon, A.M.; Das, D.; Ingraham, H.A.; Fletterick, R.J. Structure of Liver Receptor Homolog-1 (NR5A2) with PIP 3 Hormone Bound in the Ligand Binding Pocket. J. Struct. Biol. 2015, 192, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Blind, R.D. Disentangling Biological Signaling Networks by Dynamic Coupling of Signaling Lipids to Modifying Enzymes. Adv. Biol. Regul. 2014, 54, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, G.; Ortlund, E.A.; Tyeryar, K.R.; Mousley, C.J.; Ile, K.E.; Garrett, T.A.; Ren, J.; Woolls, M.J.; Raetz, C.R.H.; Redinbo, M.R.; et al. Functional Anatomy of Phospholipid Binding and Regulation of Phosphoinositide Homeostasis by Proteins of the Sec14 Superfamily. Mol. Cell 2008, 29, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ghosh, R.; Tripathi, A.; Lönnfors, M.; Somerharju, P.; Bankaitis, V.A. Two-Ligand Priming Mechanism for Potentiated Phosphoinositide Synthesis Is an Evolutionarily Conserved Feature of Sec14-Like Phosphatidylinositol and Phosphatidylcholine Exchange Proteins. Mol. Biol. Cell 2016, 27, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Ile, K.E.; Nile, A.H.; Ren, J.; Ghosh, R.; Schaaf, G. Thoughts on Sec14-like Nanoreactors and Phosphoinositide Signaling. Adv. Biol. Regul. 2012, 52, 115–121. [Google Scholar] [CrossRef]
- Ile, K.E.; Schaaf, G.; Bankaitis, V.A. Phosphatidylinositol Transfer Proteins and Cellular Nanoreactors for Lipid Signaling. Nat. Chem. Biol. 2006, 2, 576–583. [Google Scholar] [CrossRef]
- Hoffman, A.M.; Chen, Q.; Zheng, T.; Nicchitta, C.V. Heterogeneous Translational Landscape of the Endoplasmic Reticulum Revealed by Ribosome Proximity Labeling and Transcriptome Analysis. J. Biol. Chem. 2019, 294, 8942–8958. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Diversity and Selectivity in MRNA Translation on the Endoplasmic Reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Primary Role for Endoplasmic Reticulum-Bound Ribosomes in Cellular Translation Identified by Ribosome Profiling. J. Biol. Chem. 2012, 287, 5518–5527. [Google Scholar] [CrossRef]
- Val, P.; Lefrançois-Martinez, A.M.; Veyssière, G.; Martinez, A. SF-1 a Key Player in the Development and Differentiation of Steroidogenic Tissues. Nucl. Recept. 2003, 1, 8. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, E.S.; Stivison, E.A.; Blind, R.D. SF-1 Induces Nuclear PIP2. Biomolecules 2023, 13, 1509. https://doi.org/10.3390/biom13101509
Chi ES, Stivison EA, Blind RD. SF-1 Induces Nuclear PIP2. Biomolecules. 2023; 13(10):1509. https://doi.org/10.3390/biom13101509
Chicago/Turabian StyleChi, Ethan S., Elizabeth A. Stivison, and Raymond D. Blind. 2023. "SF-1 Induces Nuclear PIP2" Biomolecules 13, no. 10: 1509. https://doi.org/10.3390/biom13101509
APA StyleChi, E. S., Stivison, E. A., & Blind, R. D. (2023). SF-1 Induces Nuclear PIP2. Biomolecules, 13(10), 1509. https://doi.org/10.3390/biom13101509