


G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Transfections
2.3. β-Arrestin2 Recruitment BRET Assay
2.4. LYN BRET Receptor Internalization Assay
2.5. Go Heterotrimer Activation BRET Assay
2.6. cAMP CAMYEL BRET Assay
2.7. GRK2-GFP Recruitment BRET Assay
2.8. Intramolecular β-Arrestin2 FlAsH Assays
2.9. [3H]-Sulpiride Intact Cell Binding Assays
2.10. Western Blotting
2.11. Data Analysis and Statistics
3. Results
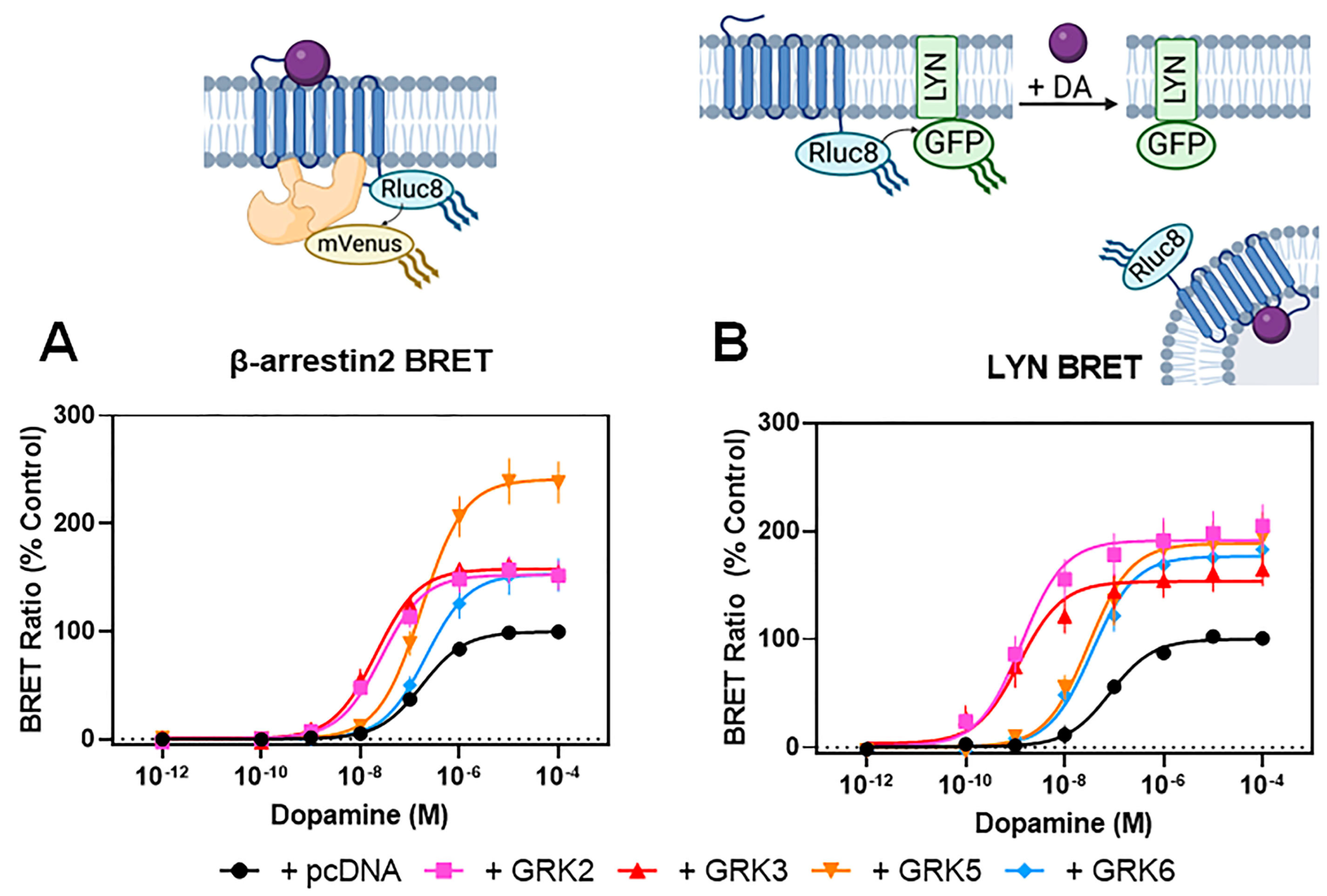
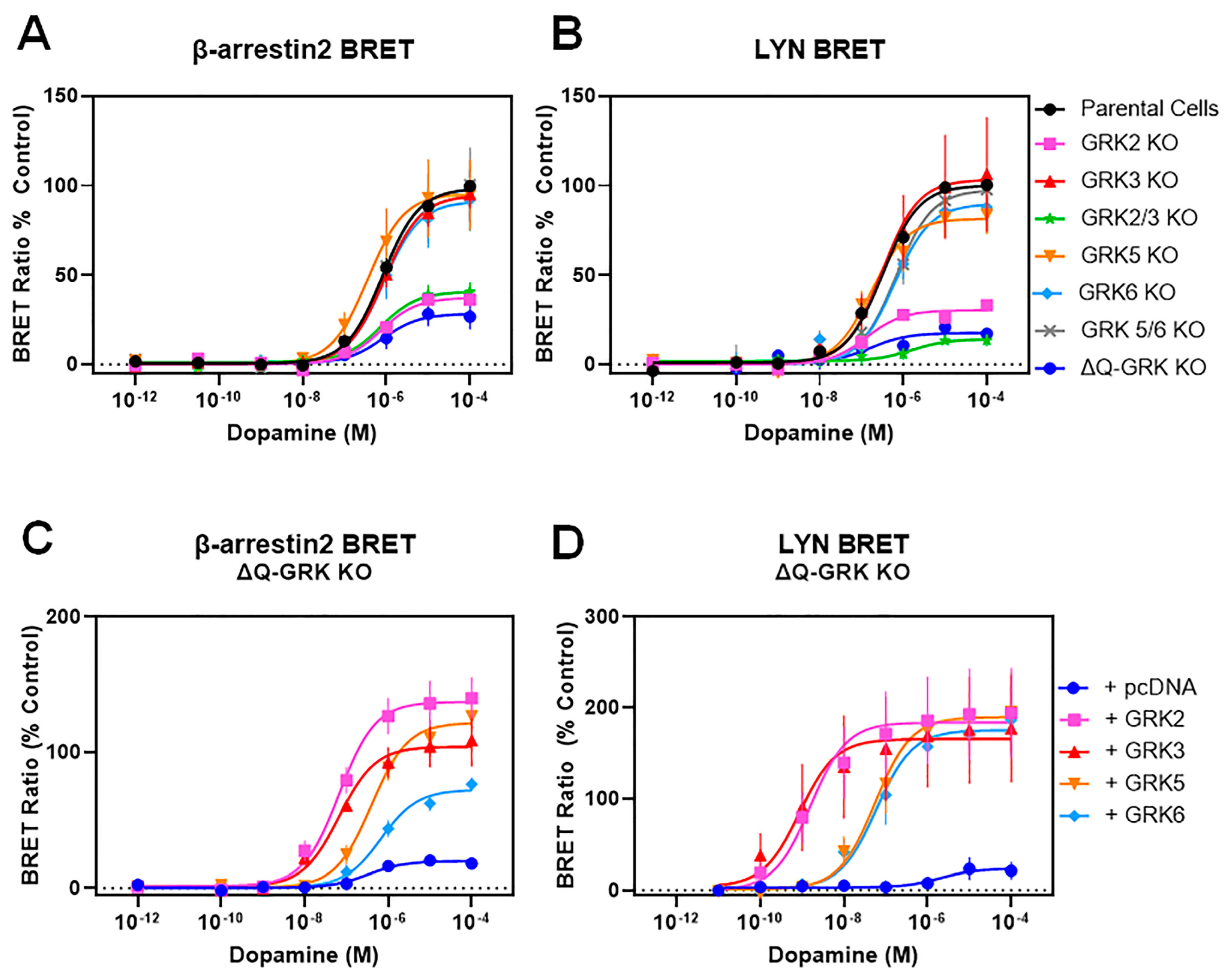
3.1. GRKs Play an Essential, Isoform-Specific Role in Modulating Interactions between β-Arrestin2 and the D2R
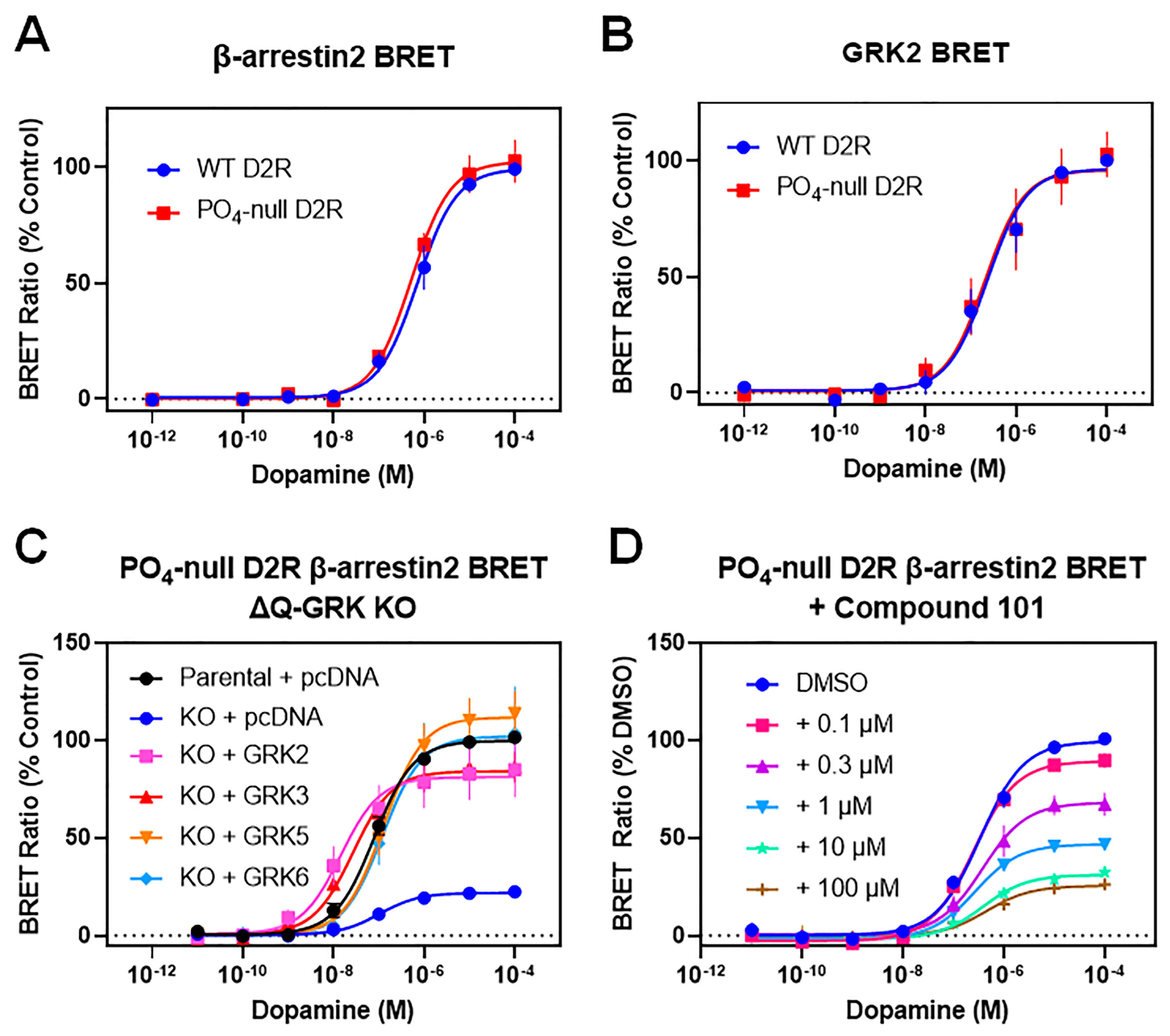
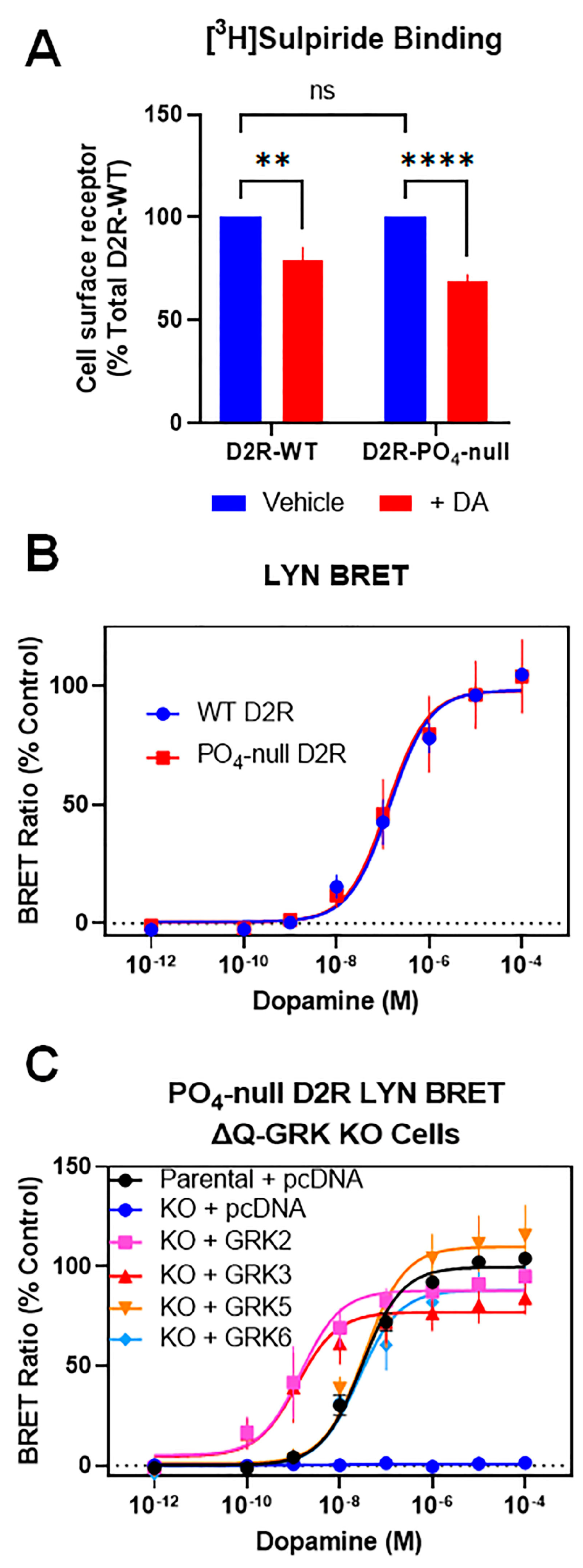
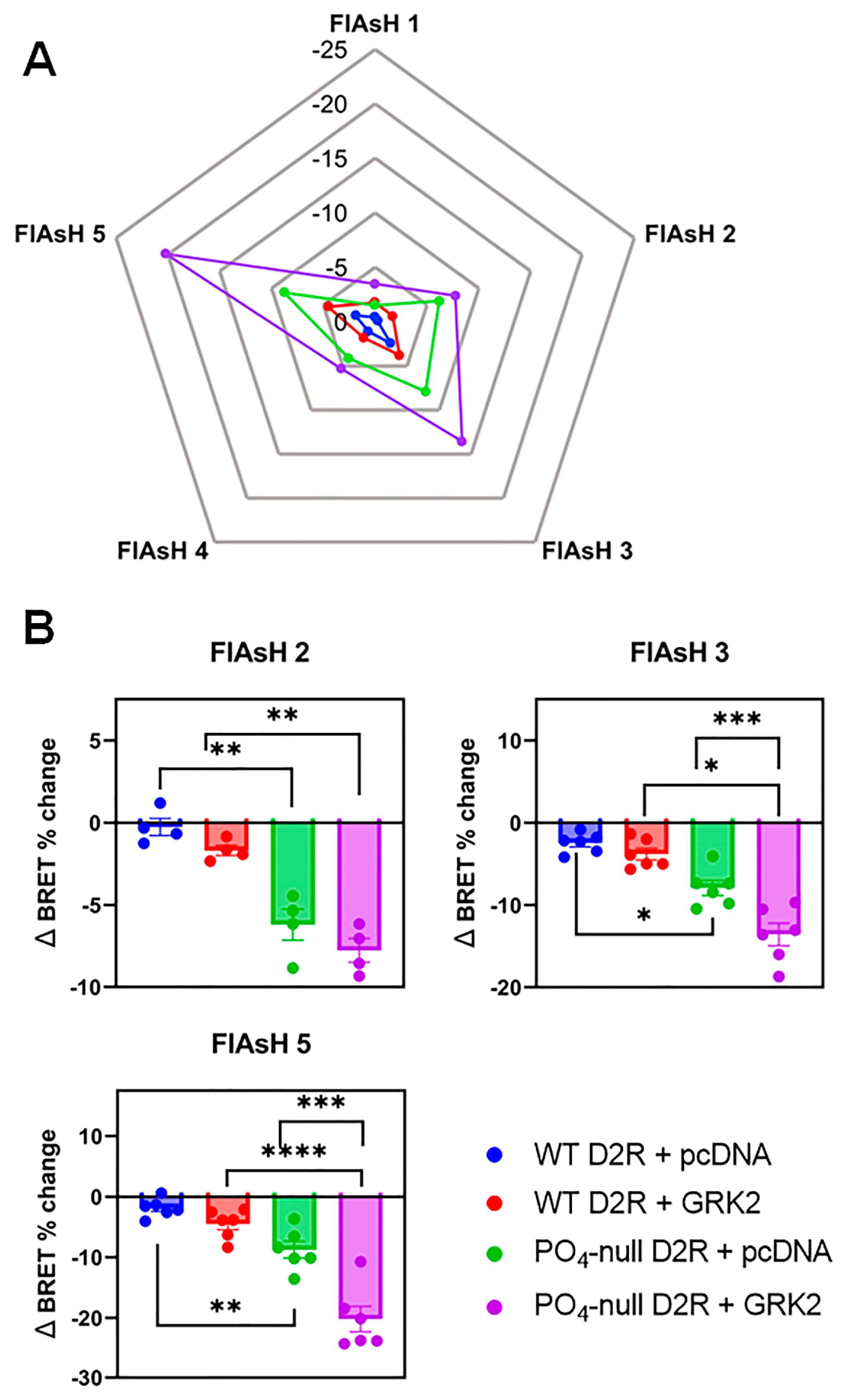
3.2. GRKs Can Enhance β-Arrestin2 Recruitment to the D2R and Receptor Internalization in the Absence of D2R Phosphorylation

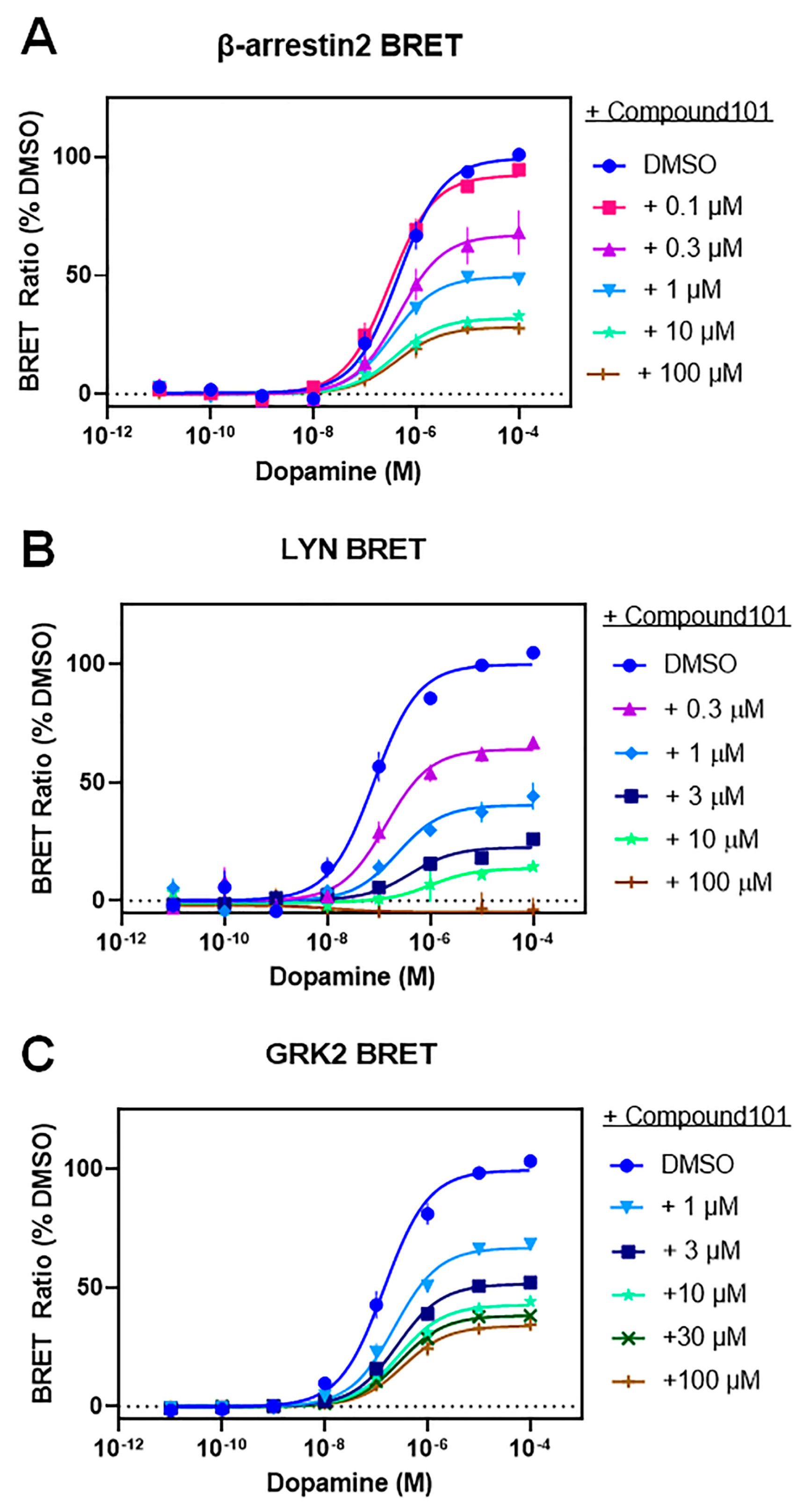
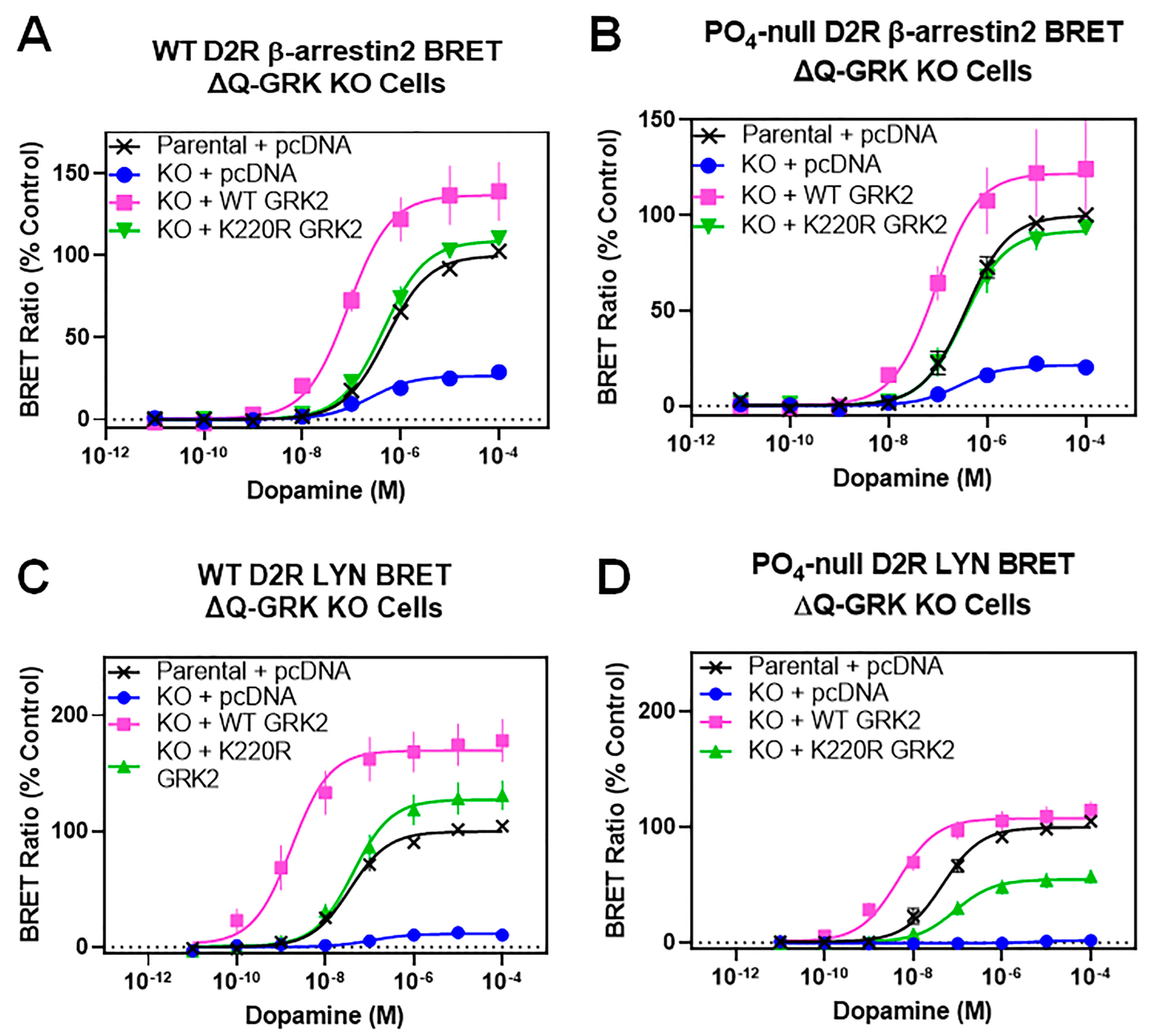
3.3. A Catalytically Inactivating Mutation of GRK2 Reveals Unique Mechanisms of GRK2-Mediated Enhancement of D2R–β-Arrestin2 Interactions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Sibley, D.R.; Monsma, F.J., Jr. Molecular biology of dopamine receptors. Trends Pharmacol. Sci. 1992, 13, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L. Historical Perspective of the G Protein-Coupled Receptor Kinase Family. Cells 2021, 10, 555. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Wang, J.K.; Bauer, B.; Townshend, R.J.L.; Hollingsworth, S.A.; Olivieri, J.E.; Xu, H.E.; Sommer, M.E.; Dror, R.O. Molecular mechanism of GPCR-mediated arrestin activation. Nature 2018, 557, 452–456. [Google Scholar] [CrossRef]
- Eichel, K.; Jullié, D.; Barsi-Rhyne, B.; Latorraca, N.R.; Masureel, M.; Sibarita, J.B.; Dror, R.O.; von Zastrow, M. Catalytic activation of β-arrestin by GPCRs. Nature 2018, 557, 381–386. [Google Scholar] [CrossRef]
- Maharana, J.; Banerjee, R.; Yadav, M.K.; Sarma, P.; Shukla, A.K. Emerging structural insights into GPCR-β-arrestin interaction and functional outcomes. Curr. Opin. Struct. Biol. 2022, 75, 102406. [Google Scholar] [CrossRef]
- Maharana, J.; Sarma, P.; Yadav, M.K.; Saha, S.; Singh, V.; Saha, S.; Chami, M.; Banerjee, R.; Shukla, A.K. Structural snapshots uncover a key phosphorylation motif in GPCRs driving β-arrestin activation. Mol. Cell 2023, 83, 2091–2107.e2097. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. The structural basis of the arrestin binding to GPCRs. Mol. Cell. Endocrinol. 2019, 484, 34–41. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. Seven-transmembrane receptor signaling through beta-arrestin. Sci. STKE 2005, 2005, cm10. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.S.; Tian, X.; Benovic, J.L. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 2014, 27, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Moritz, A.E.; Madaras, N.S.; Rankin, M.L.; Inbody, L.R.; Sibley, D.R. Delineation of G Protein-Coupled Receptor Kinase Phosphorylation Sites within the D(1) Dopamine Receptor and Their Roles in Modulating β-Arrestin Binding and Activation. Int. J. Mol. Sci. 2023, 24, 6599. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef]
- Namkung, Y.; Dipace, C.; Urizar, E.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem. 2009, 284, 34103–34115. [Google Scholar] [CrossRef]
- Namkung, Y.; Dipace, C.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem. 2009, 284, 15038–15051. [Google Scholar] [CrossRef]
- Cho, D.; Zheng, M.; Min, C.; Ma, L.; Kurose, H.; Park, J.H.; Kim, K.M. Agonist-induced endocytosis and receptor phosphorylation mediate resensitization of dopamine D(2) receptors. Mol. Endocrinol. 2010, 24, 574–586. [Google Scholar] [CrossRef]
- Cho, D.I.; Zheng, M.; Min, C.; Kwon, K.J.; Shin, C.Y.; Choi, H.K.; Kim, K.M. ARF6 and GASP-1 are post-endocytic sorting proteins selectively involved in the intracellular trafficking of dopamine D2 receptors mediated by GRK and PKC in transfected cells. Br. J. Pharmacol. 2013, 168, 1355–1374. [Google Scholar] [CrossRef]
- Kim, K.M.; Valenzano, K.J.; Robinson, S.R.; Yao, W.D.; Barak, L.S.; Caron, M.G. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J. Biol. Chem. 2001, 276, 37409–37414. [Google Scholar] [CrossRef]
- Pack, T.F.; Orlen, M.I.; Ray, C.; Peterson, S.M.; Caron, M.G. The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J. Biol. Chem. 2018, 293, 6161–6171. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.; Keen, A.C.; Mark, H.; Dasgupta, P.; Javitch, J.A.; Canals, M.; Schulz, S.; Robert Lane, J. New phosphosite-specific antibodies to unravel the role of GRK phosphorylation in dopamine D(2) receptor regulation and signaling. Sci. Rep. 2021, 11, 8288. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Homan, K.T.; Vishnivetskiy, S.A.; Manglik, A.; Tesmer, J.J.; Gurevich, V.V.; Gurevich, E.V. G Protein-coupled Receptor Kinases of the GRK4 Protein Subfamily Phosphorylate Inactive G Protein-coupled Receptors (GPCRs). J. Biol. Chem. 2015, 290, 10775–10790. [Google Scholar] [CrossRef]
- Kim, K.M. Unveiling the Differences in Signaling and Regulatory Mechanisms between Dopamine D(2) and D(3) Receptors and Their Impact on Behavioral Sensitization. Int. J. Mol. Sci. 2023, 24, 6742. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Bohn, L.M.; Sotnikova, T.D.; Cyr, M.; Laakso, A.; Macrae, A.D.; Torres, G.E.; Kim, K.M.; Lefkowitz, R.J.; Caron, M.G.; et al. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron 2003, 38, 291–303. [Google Scholar] [CrossRef]
- Managò, F.; Espinoza, S.; Salahpour, A.; Sotnikova, T.D.; Caron, M.G.; Premont, R.T.; Gainetdinov, R.R. The role of GRK6 in animal models of Parkinson’s disease and L-DOPA treatment. Sci. Rep. 2012, 2, 301. [Google Scholar] [CrossRef]
- Drube, J.; Haider, R.S.; Matthees, E.S.F.; Reichel, M.; Zeiner, J.; Fritzwanker, S.; Ziegler, C.; Barz, S.; Klement, L.; Filor, J.; et al. GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat. Commun. 2022, 13, 540. [Google Scholar] [CrossRef]
- Haider, R.S.; Matthees, E.S.F.; Drube, J.; Reichel, M.; Zabel, U.; Inoue, A.; Chevigné, A.; Krasel, C.; Deupi, X.; Hoffmann, C. β-arrestin1 and 2 exhibit distinct phosphorylation-dependent conformations when coupling to the same GPCR in living cells. Nat. Commun. 2022, 13, 5638. [Google Scholar] [CrossRef]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef]
- Hoffmann, C.; Gaietta, G.; Zürn, A.; Adams, S.R.; Terrillon, S.; Ellisman, M.H.; Tsien, R.Y.; Lohse, M.J. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protoc. 2010, 5, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Skinbjerg, M.; Ariano, M.A.; Thorsell, A.; Heilig, M.; Halldin, C.; Innis, R.B.; Sibley, D.R. Arrestin3 mediates D(2) dopamine receptor internalization. Synapse 2009, 63, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.C.; Donthamsetti, P.; Lambert, N.A.; Javitch, J.A.; Neve, K.A. Mutation of three residues in the third intracellular loop of the dopamine D2 receptor creates an internalization-defective receptor. J. Biol. Chem. 2014, 289, 33663–33675. [Google Scholar] [CrossRef]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [PubMed]
- Namkung, Y.; Le Gouill, C.; Lukashova, V.; Kobayashi, H.; Hogue, M.; Khoury, E.; Song, M.; Bouvier, M.; Laporte, S.A. Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 2016, 7, 12178. [Google Scholar] [CrossRef]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef]
- Eiger, D.S.; Smith, J.S.; Shi, T.; Stepniewski, T.M.; Tsai, C.F.; Honeycutt, C.; Boldizsar, N.; Gardner, J.; Nicora, C.D.; Moghieb, A.M.; et al. Phosphorylation barcodes direct biased chemokine signaling at CXCR3. Cell Chem. Biol. 2023, 30, 362–382.e368. [Google Scholar] [CrossRef]
- Namkung, Y.; Sibley, D.R. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J. Biol. Chem. 2004, 279, 49533–49541. [Google Scholar] [CrossRef]
- Xiao, K.; Shenoy, S.K.; Nobles, K.; Lefkowitz, R.J. Activation-dependent conformational changes in {beta}-arrestin 2. J. Biol. Chem. 2004, 279, 55744–55753. [Google Scholar] [CrossRef]
- Lee, M.H.; Appleton, K.M.; Strungs, E.G.; Kwon, J.Y.; Morinelli, T.A.; Peterson, Y.K.; Laporte, S.A.; Luttrell, L.M. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 2016, 531, 665–668. [Google Scholar] [CrossRef]
- Kong, G.; Penn, R.; Benovic, J.L. A beta-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the beta 2-adrenergic receptor. J. Biol. Chem. 1994, 269, 13084–13087. [Google Scholar] [CrossRef]
- Dale, L.B.; Bhattacharya, M.; Anborgh, P.H.; Murdoch, B.; Bhatia, M.; Nakanishi, S.; Ferguson, S.S. G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J. Biol. Chem. 2000, 275, 38213–38220. [Google Scholar] [CrossRef] [PubMed]
- Reichel, M.; Weitzel, V.; Klement, L.; Hoffmann, C.; Drube, J. Suitability of GRK Antibodies for Individual Detection and Quantification of GRK Isoforms in Western Blots. Int. J. Mol. Sci. 2022, 23, 1195. [Google Scholar] [CrossRef] [PubMed]
- Møller, T.C.; Pedersen, M.F.; van Senten, J.R.; Seiersen, S.D.; Mathiesen, J.M.; Bouvier, M.; Bräuner-Osborne, H. Dissecting the roles of GRK2 and GRK3 in μ-opioid receptor internalization and β-arrestin2 recruitment using CRISPR/Cas9-edited HEK293 cells. Sci. Rep. 2020, 10, 17395. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Soto, M.; Verma, R.K.; Willette, B.K.A.; Gonye, E.C.; Moore, A.M.; Moritz, A.E.; Boateng, C.A.; Yano, H.; Free, R.B.; Shi, L.; et al. A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci. Signal. 2020, 13, eaaw5885. [Google Scholar] [CrossRef]
- Homan, K.T.; Tesmer, J.J. Molecular basis for small molecule inhibition of G protein-coupled receptor kinases. ACS Chem. Biol. 2015, 10, 246–256. [Google Scholar] [CrossRef]
- Wess, J.; Oteng, A.B.; Rivera-Gonzalez, O.; Gurevich, E.V.; Gurevich, V.V. β-Arrestins: Structure, Function, Physiology, and Pharmacological Perspectives. Pharmacol. Rev. 2023, 75, 854–884. [Google Scholar] [CrossRef]
- Gantz, S.C.; Robinson, B.G.; Buck, D.C.; Bunzow, J.R.; Neve, R.L.; Williams, J.T.; Neve, K.A. Distinct regulation of dopamine D2S and D2L autoreceptor signaling by calcium. Elife 2015, 4, e09358. [Google Scholar] [CrossRef]
- Ågren, R.; Sahlholm, K. G protein-coupled receptor kinase-2 confers isoform-specific calcium sensitivity to dopamine D(2) receptor desensitization. FASEB J. 2021, 35, e22013. [Google Scholar] [CrossRef]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef]
- Evron, T.; Daigle, T.L.; Caron, M.G. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012, 33, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell. Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dopamine-Stimulated | +pcDNA | +GRK2 | +GRK3 | +GRK5 | +GRK6 | |
|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 190 ± 28 | 29 ± 3.3 **** | 24 ± 4.8 **** | 190 ± 19 | 230 ± 23 |
| Emax (% + pcDNA) | 100 | 150 ± 12 * | 160 ± 5.1 * | 240 ± 20 **** | 150 ± 16 * | |
| LYN BRET | EC50 (nM) | 90 ± 24 | 1.6 ± 0.5 **** | 2.1 ± 1.0 **** | 36 ± 11 | 43 ± 14 |
| Emax (% + pcDNA) | 100 | 190 ± 19 ** | 150 ± 15 * | 190 ± 23 ** | 180 ± 7.8 ** | |
| Dopamine-Stimulated | Parental HEK293 | GRK2 KO | GRK3 KO | GRK2/3 KO | GRK5 KO | GRK6 KO | GRK 5/6 KO | ΔQ-GRK KO | |
|---|---|---|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 1300 ± 340 | 1000 ± 340 | 1200 ± 410 | 810 ± 270 | 580 ± 120 | 1600 ± 530 | 1400 ± 490 | 1600 ± 570 |
| Emax (% Parental) | 100 | 38 ± 2.9 *** | 96 ± 7.7 | 41 ± 5.6 *** | 96 ± 21 | 92 ± 17 | 99 ± 20 | 29 ± 6.9 **** | |
| LYN BRET | EC50 (nM) | 380 ± 130 | 160 ± 38 | 410 ± 160 | ND | 205 ± 76 | 660 ± 110 | 800 ± 280 | ND |
| Emax (% Parental) | 100 | 31 ± 0.7 *** | 104 ± 30 | ND | 82 ± 11 | 90 ± 5.7 | 98 ± 9.3 | ND | |
| Dopamine- Stimulated | Parental + pcDNA | ΔQ-KO + pcDNA | ΔQ-KO + GRK2 | ΔQ-KO + GRK3 | ΔQ-KO + GRK5 | ΔQ-KO + GRK6 | |
|---|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 500 ± 110 | 390 ± 150 | 81 ± 37 * | 71 ± 31 ** | 460 ± 150 | 890 ± 410 |
| Emax (% Parental) | 100 | 20 ± 2.2 *** | 140 ± 16 | 105 ± 17 | 120 ± 16 | 73 ± 4.7 | |
| LYN BRET | EC50 (nM) | 91 ± 37 | ND | 2.6 ± 1.0 *** | 3.9 ± 2.1 *** | 81 ± 32 | 110 ± 67 |
| Emax (% Parental) | 100 | ND | 180 ± 47 | 170 ± 55 | 190 ± 42 | 180 ± 35 | |
| Dopamine-Stimulated | DMSO | +C-101 0.1 µM | +C-101 0.3 µM | +C-101 1 µM | +C-101 3 µM | +C-101 10 µM | +C-101 30 µM | +C-101 100 µM | |
|---|---|---|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 560 ± 170 | 330 ± 81 | 470 ± 59 | 380 ± 130 | NT | 660 ± 260 | NT | 570 ± 280 |
| Emax (% DMSO) | 100 | 92 ± 1.2 | 67 ± 8.4 **** | 50 ± 2.0 **** | NT | 33 ± 1.2 **** | NT | 28 ± 1.7 **** | |
| LYN BRET | EC50 (nM) | 85 ± 25 | NT | 160 ± 49 | 300 ± 120 | 580 ± 360 | 250 ± 130 | NT | ND |
| Emax (% DMSO) | 100 | NT | 64 ± 2.6 ** | 41 ± 4.6 **** | 23 ± 0.8 **** | 23 ± 9.5 **** | NT | ND | |
| GRK2 BRET | EC50 (nM) | 170 ± 56 | NT | NT | 260 ± 98 | 280 ± 76 | 300 ± 85 | 300 ± 47 | 370 ± 88 |
| Emax (% DMSO) | 100 | NT | NT | 67 ± 2.4 **** | 52 ± 1.1 **** | 43 ± 1.3 **** | 38 ± 2.4 **** | 34.0 ± 2.2 **** | |
| Dopamine- Stimulated | Parental + pcDNA | ΔQ-KO + pcDNA | ΔQ-KO + GRK2 | ΔQ-KO + GRK3 | ΔQ-KO + GRK5 | ΔQ-KO + GRK6 | |
|---|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 99 ± 29 | 106 ± 21 | 20 ± 6.5 *** | 28 ± 7.7 * | 160 ± 39 | 120 ± 8.6 |
| Emax (% Parental) | 100 | 22 ± 1.1 **** | 81 ± 13 | 85 ± 8.3 | 113 ± 12 | 102 ± 23 | |
| LYN BRET | EC50 (nM) | 34 ± 8.0 | ND | 2.5 ± 2.0 * | 2.6 ± 2.2 * | 53 ± 38 | 49 ± 33 |
| Emax (% Parental) | 100 | ND | 88 ± 3.5 | 77 ± 7.7 | 103 ± 13 | 90 ± 8.5 | |
| Dopamine-Stimulated | DMSO | +C-101 0.1 µM | +C-101 0.3 µM | +C-101 1 µM | +C-101 10 µM | +C-101 100 µM | |
|---|---|---|---|---|---|---|---|
| β-arrestin2 BRET | EC50 (nM) | 410 ± 120 | 250 ± 21 | 440 ± 140 | 280 ± 46 | 430 ± 140 | 580 ± 230 |
| Emax (% DMSO) | 100 | 92 ± 3.1 | 68 ± 5.0 **** | 49 ± 1.6 **** | 33 ± 1.9 **** | 26 ± 4.6 **** | |
| Dopamine- Stimulated | Parental + pcDNA | ΔQ-KO + pcDNA | ΔQ-KO + WT GRK2 | ΔQ-KO + K220R GRK2 | ||
|---|---|---|---|---|---|---|
| β-arrestin2 BRET | WT D2R | EC50 (nM) | 530 ± 78 | 370 ± 210 | 91 ± 34 * | 480 ± 130 |
| Emax (% Parental) | 100 | 23 ± 4.1 **** | 135 ± 13 * | 101 ± 8.6 | ||
| PO4-null D2R | EC50 (nM) | 400 ± 130 | 270 ± 14 | 88 ± 22 * | 400 ± 140 | |
| Emax (% Parental) | 100 | 21 ± 0.8 ** | 122 ± 24 | 92 ± 3.1 | ||
| LYN BRET | WT D2R | EC50 (nM) | 41 ± 10 | ND | 2.4 ± 0.8 ** | 48 ± 13 |
| Emax (% Parental) | 100 | ND | 170 ± 18 ** | 130 ± 13 | ||
| PO4-null D2R | EC50 (nM) | 53 ± 13 | ND | 4.9 ± 1.3 ** | 110 ± 25 | |
| Emax (% Parental) | 100 | ND | 107 ± 8.3 | 55 ± 6.0 *** | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Soto, M.; Boldizsar, N.M.; Schardien, K.A.; Madaras, N.S.; Willette, B.K.A.; Inbody, L.R.; Dasaro, C.; Moritz, A.E.; Drube, J.; Haider, R.S.; et al. G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation. Biomolecules 2023, 13, 1552. https://doi.org/10.3390/biom13101552
Sánchez-Soto M, Boldizsar NM, Schardien KA, Madaras NS, Willette BKA, Inbody LR, Dasaro C, Moritz AE, Drube J, Haider RS, et al. G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation. Biomolecules. 2023; 13(10):1552. https://doi.org/10.3390/biom13101552
Chicago/Turabian StyleSánchez-Soto, Marta, Noelia M. Boldizsar, Kayla A. Schardien, Nora S. Madaras, Blair K. A. Willette, Laura R. Inbody, Christopher Dasaro, Amy E. Moritz, Julia Drube, Raphael S. Haider, and et al. 2023. "G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation" Biomolecules 13, no. 10: 1552. https://doi.org/10.3390/biom13101552
APA StyleSánchez-Soto, M., Boldizsar, N. M., Schardien, K. A., Madaras, N. S., Willette, B. K. A., Inbody, L. R., Dasaro, C., Moritz, A. E., Drube, J., Haider, R. S., Free, R. B., Hoffman, C., & Sibley, D. R. (2023). G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation. Biomolecules, 13(10), 1552. https://doi.org/10.3390/biom13101552