
Proteome Profiling of the Dystrophic mdx Mice Diaphragm
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. Protein Isolation
2.3. Total Protein Measurement
2.4. Protein Digestion
2.5. TMT Labeling
2.6. Reversed-Phase Peptide Fractionation of TMT-Labeled Peptides
2.7. Mass Spectrometry
2.8. Data Analysis
2.8.1. LF Analysis
2.8.2. TMT Analysis
2.9. Western Blot
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Histological Analysis
2.12. RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR (qRT-PCR)
2.13. Statistical Analysis
3. Results
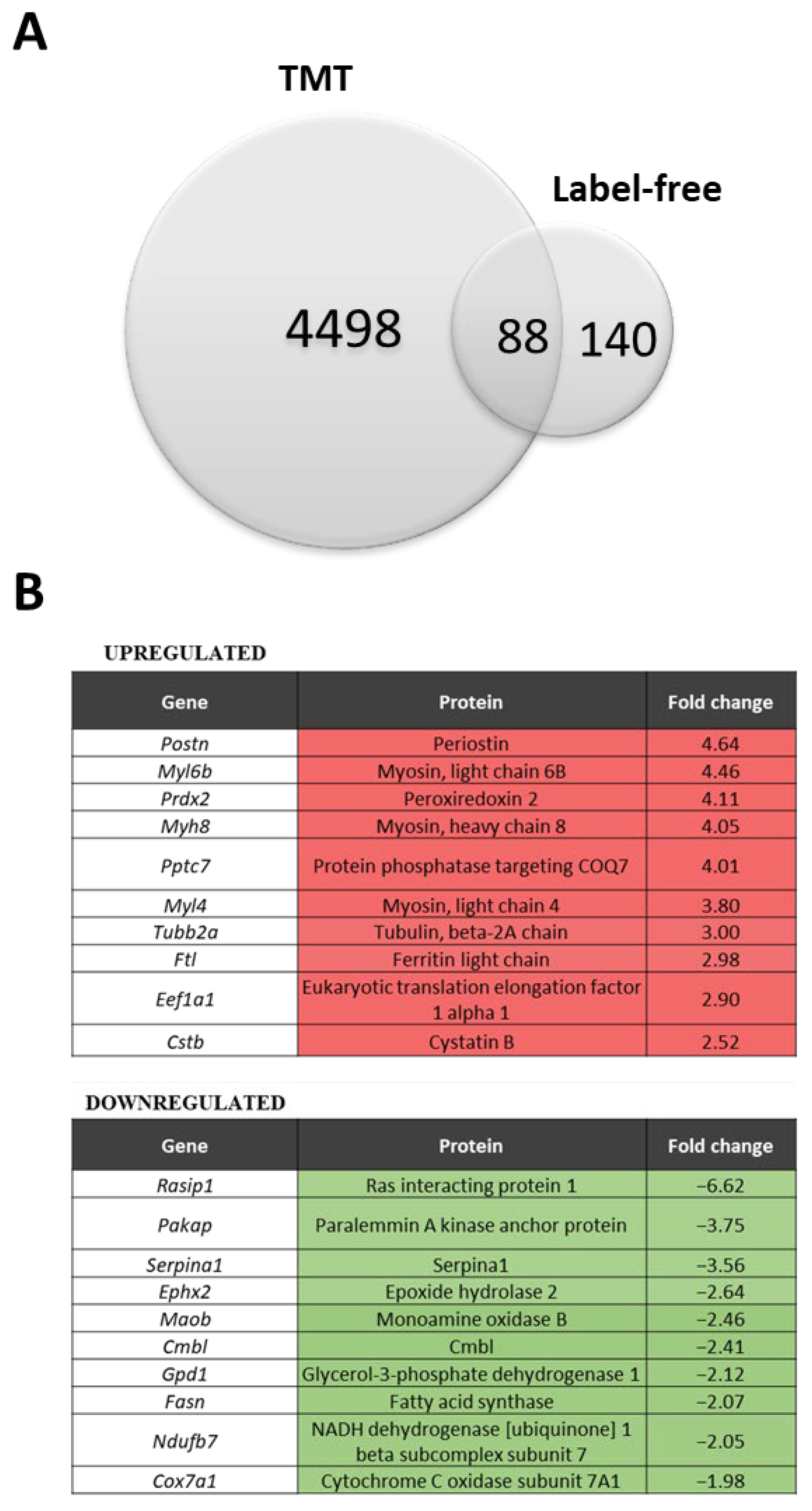
3.1. Preliminary Comparative Results from Both the Label-Free (LF) Method and the Tandem Mass Tag (TMT) Approach
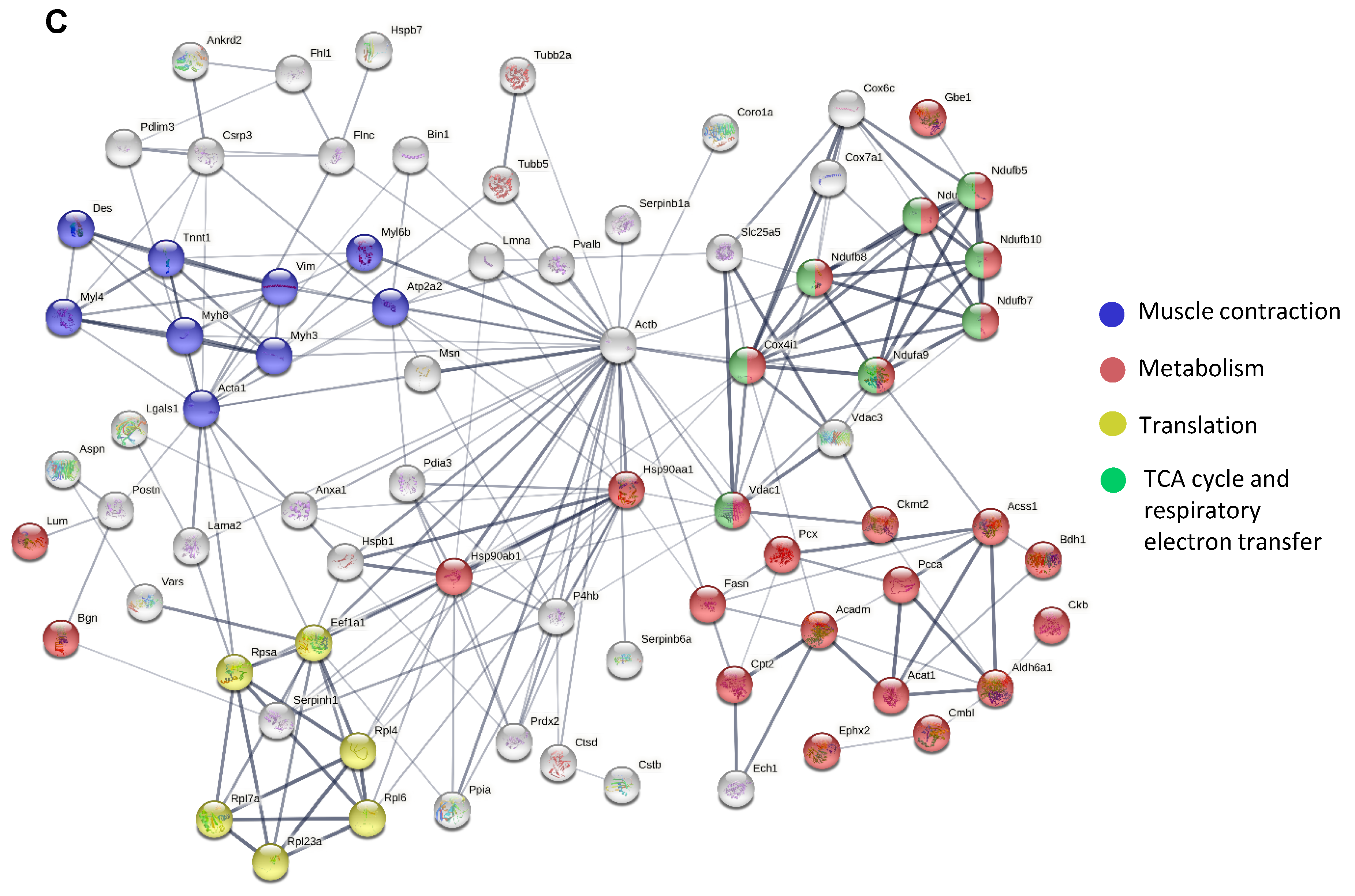
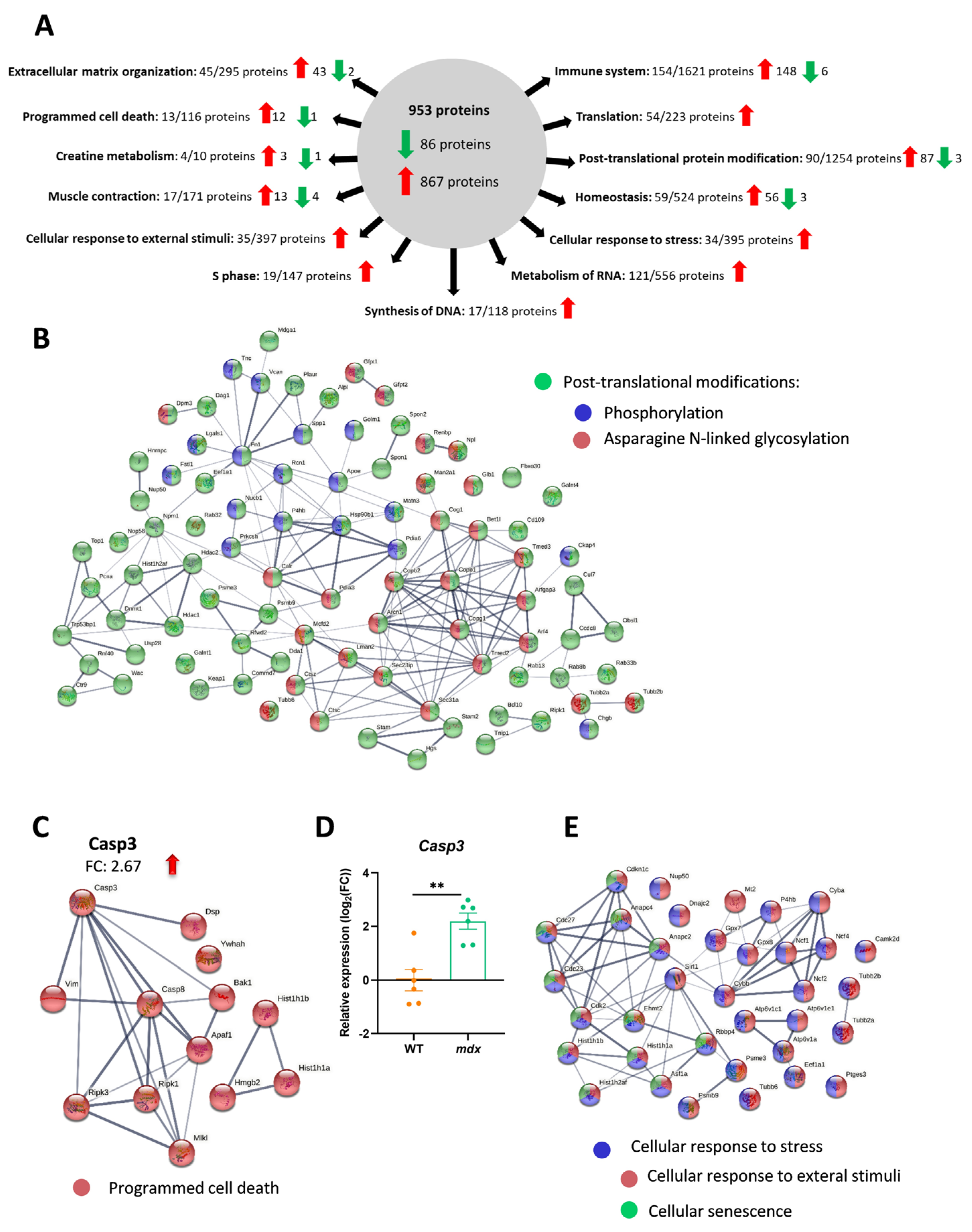
3.2. TMT Analysis Revealed Proteins with Increased Rather Than Decreased Abundance in Dystrophic Animals
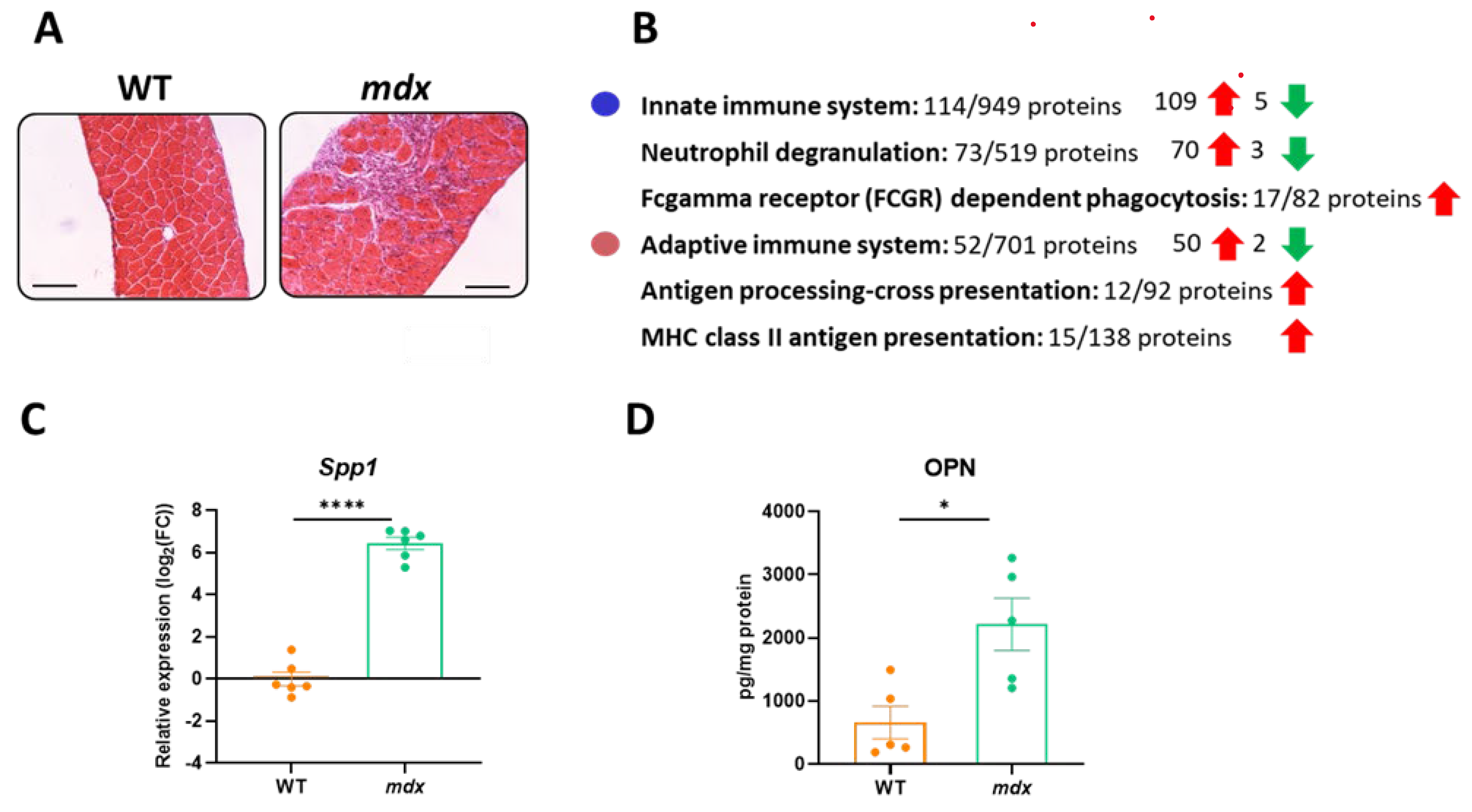
3.3. Immune-System-Related Pathways Are Strongly Enriched in the Diaphragm Muscle of Dystrophic Animals
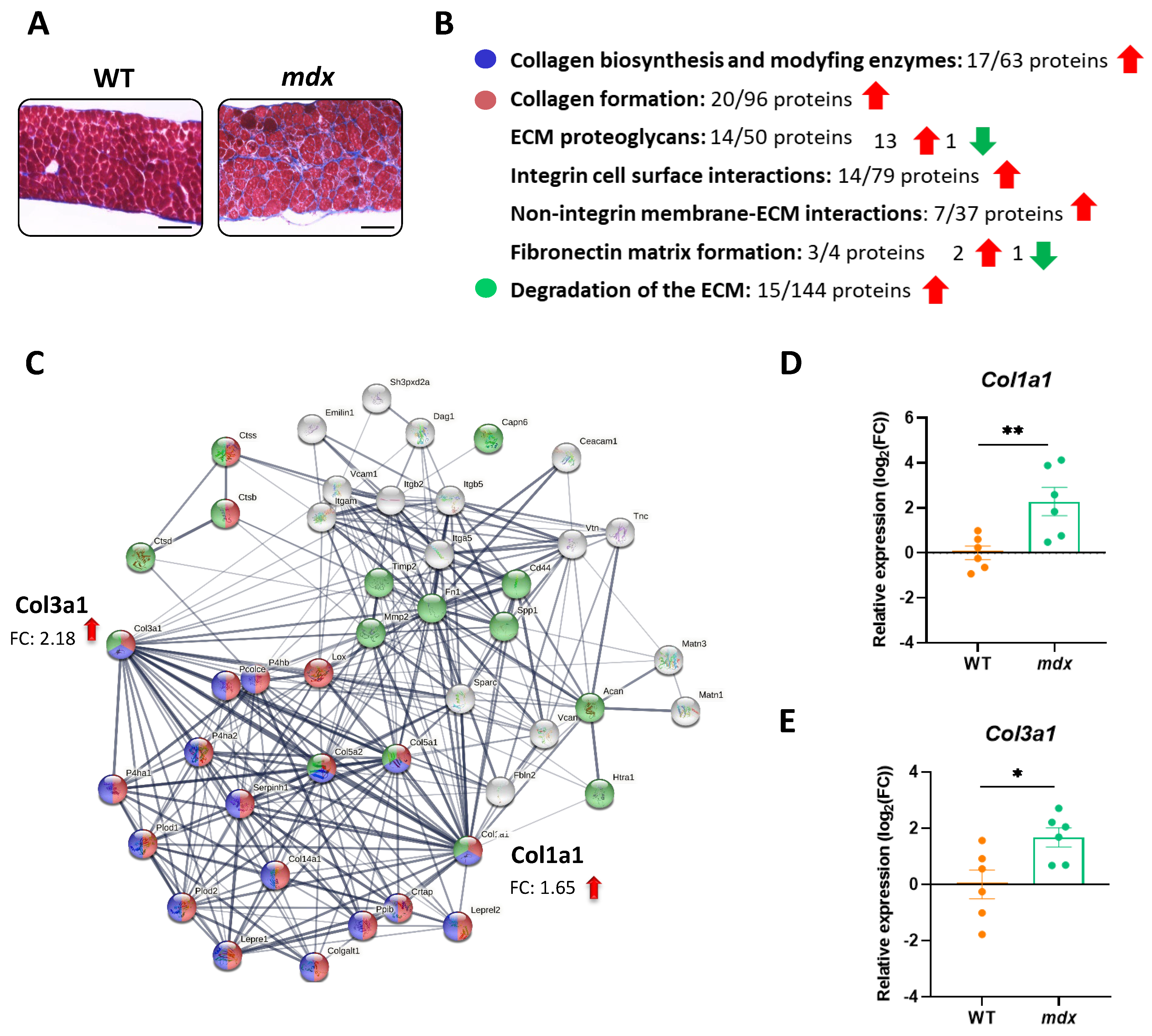
3.4. The Organization of the Extracellular Matrix (ECM) Is Disturbed in the Diaphragm of Dystrophic Mice
3.5. The Lack of Dystrophin Affects Protein Metabolism and Processes Related to the Cellular Response to Stress
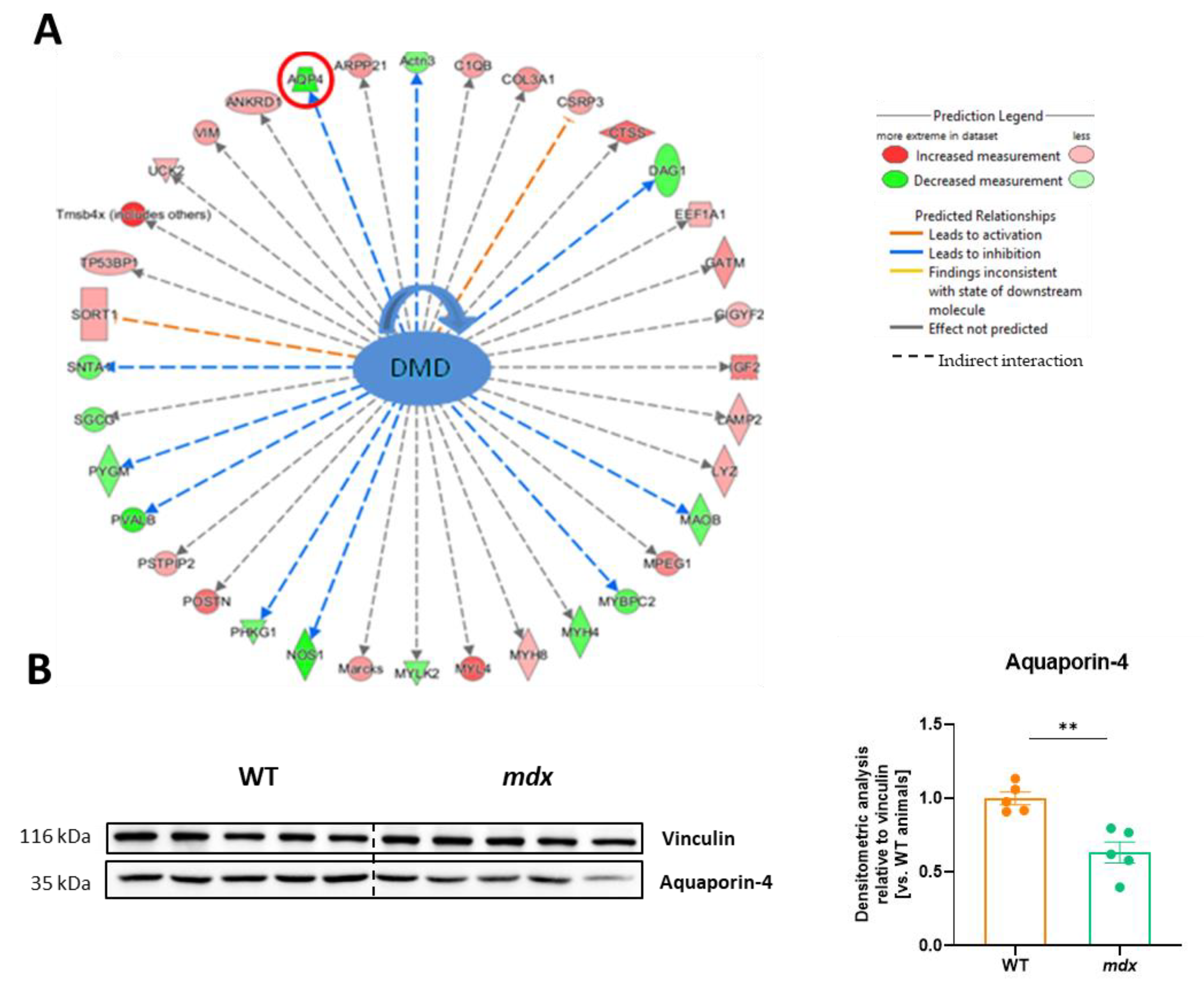
3.6. The Lack of Dystrophin Results in Significant Changes in Several Proteins, including the Water Channel Protein, Aquaporin 4 (AQP4)
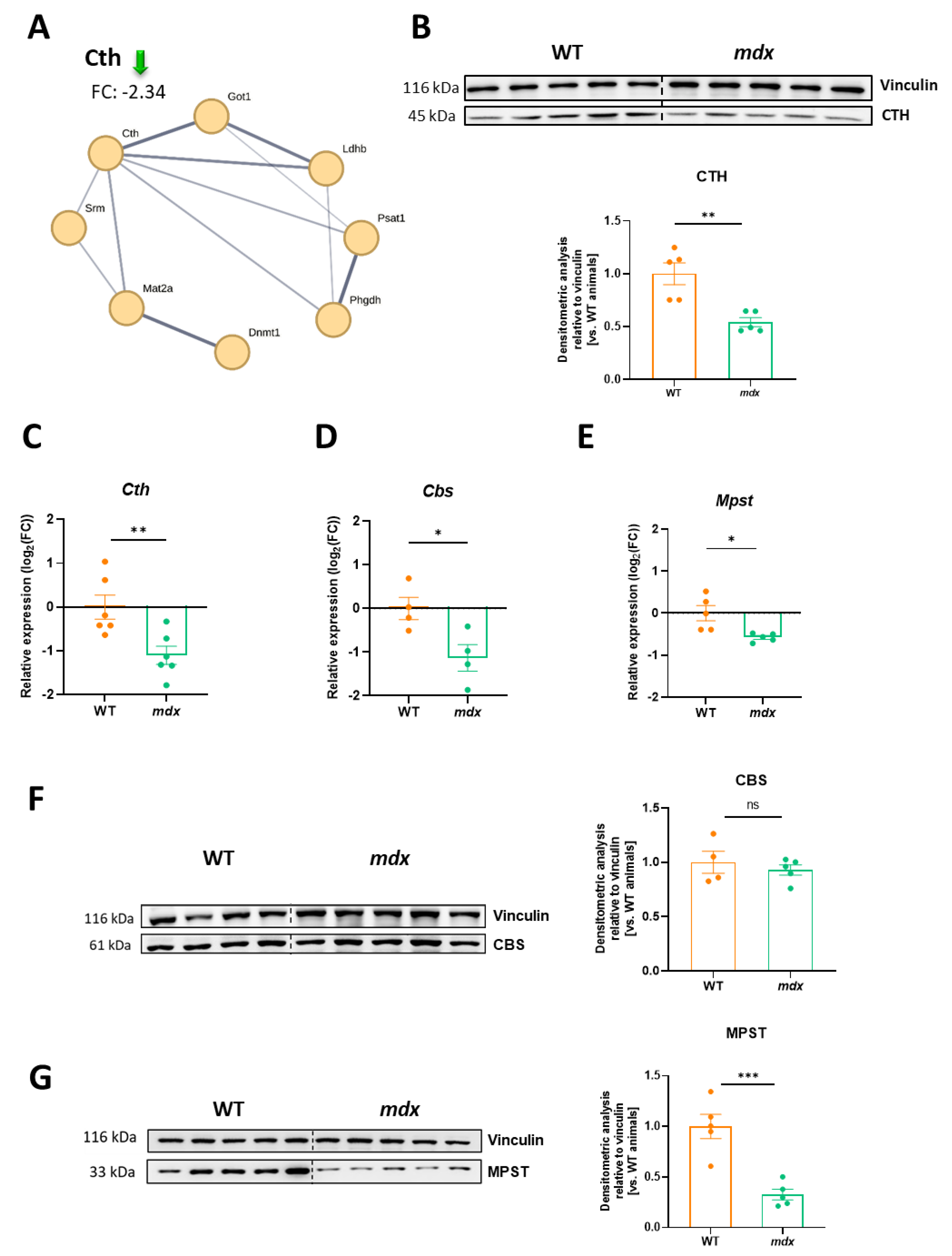
3.7. Decreased Expression of H2S-Generating Enzymes Is a Hallmark of DMD
4. Discussion
5. Conclusions
6. Study Limitations and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flanigan, K.M. The Muscular Dystrophies. Semin. Neurol. 2012, 32, 255–263. [Google Scholar] [CrossRef]
- Emery, A.E. The Muscular Dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Mercuri, E. Evidence-Based Care in Duchenne Muscular Dystrophy. Lancet Neurol. 2018, 17, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Muscle and Cardiac Therapeutic Strategies for Duchenne Muscular Dystrophy: Past, Present, and Future. Pharmacol. Rep. 2020, 72, 1227–1263. [Google Scholar] [CrossRef]
- Podkalicka, P.; Myszka, M.; Dulak, J.; Łoboda, A. Molecular mechanisms of Duchenne muscular dystrophy and new therapeutic strategies. Postepy Biochem. 2022, 68, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; McNally, E.M. Mechanisms of Muscle Weakness in Muscular Dystrophy. J. Gen. Physiol. 2010, 136, 29–34. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Respiratory, Cardiac, Bone Health, and Orthopaedic Management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Broomfield, J.; Hill, M.; Guglieri, M.; Crowther, M.; Abrams, K. Life Expectancy in Duchenne Muscular Dystrophy: Reproduced Individual Patient Data Meta-Analysis. Neurology 2021, 97, e2304–e2314. [Google Scholar] [CrossRef] [PubMed]
- Florczyk-Soluch, U.; Polak, K.; Dulak, J. The Multifaceted View of Heart Problem in Duchenne Muscular Dystrophy. Cell. Mol. Life Sci. 2021, 78, 5447–5468. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X Chromosome-Linked Muscular Dystrophy (Mdx) in the Mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The Molecular Basis of Muscular Dystrophy in the Mdx Mouse: A Point Mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Dangain, J.; Vrbova, G. Muscle Development in Mdx Mutant Mice. Muscle Nerve 1984, 7, 700–704. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal Models of Duchenne Muscular Dystrophy: From Basic Mechanisms to Gene Therapy. Dis. Models Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef]
- Cullen, M.J.; Jaros, E. Ultrastructure of the Skeletal Muscle in the X Chromosome-Linked Dystrophic (Mdx) Mouse. Comparison with Duchenne Muscular Dystrophy. Acta Neuropathol. 1988, 77, 69–81. [Google Scholar] [CrossRef]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The Mdx Mouse Diaphragm Reproduces the Degenerative Changes of Duchenne Muscular Dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Coirault, C.; Pignol, B.; Cooper, R.N.; Butler-Browne, G.; Chabrier, P.-E.; Lecarpentier, Y. Severe Muscle Dysfunction Precedes Collagen Tissue Proliferation in Mdx Mouse Diaphragm. J. Appl. Physiol. 2003, 94, 1744–1750. [Google Scholar] [CrossRef]
- Mhandire, D.Z.; Burns, D.P.; Roger, A.L.; O’Halloran, K.D.; ElMallah, M.K. Breathing in Duchenne Muscular Dystrophy: Translation to Therapy. J. Physiol. 2022, 600, 3465–3482. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Extracellular Matrix Proteomics: The Mdx-4cv Mouse Diaphragm as a Surrogate for Studying Myofibrosis in Dystrophinopathy. Biomolecules 2023, 13, 1108. [Google Scholar] [CrossRef] [PubMed]
- Kaziród, K.; Myszka, M.; Dulak, J.; Łoboda, A. Hydrogen Sulfide as a Therapeutic Option for the Treatment of Duchenne Muscular Dystrophy and Other Muscle-Related Diseases. Cell. Mol. Life Sci. 2022, 79, 608. [Google Scholar] [CrossRef] [PubMed]
- Myszka, M.; Mucha, O.; Podkalicka, P.; Waśniowska, U.; Dulak, J.; Łoboda, A. Sodium Hydrosulfide Moderately Alleviates the Hallmark Symptoms of Duchenne Muscular Dystrophy in Mdx Mice. Eur. J. Pharmacol. 2023, 955, 175928. [Google Scholar] [CrossRef]
- Panza, E.; Vellecco, V.; Iannotti, F.A.; Paris, D.; Manzo, O.L.; Smimmo, M.; Mitilini, N.; Boscaino, A.; de Dominicis, G.; Bucci, M.; et al. Duchenne’s Muscular Dystrophy Involves a Defective Transsulfuration Pathway Activity. Redox Biol. 2021, 45, 102040. [Google Scholar] [CrossRef]
- Saclier, M.; Ben Larbi, S.; My Ly, H.; Moulin, E.; Mounier, R.; Chazaud, B.; Juban, G. Interplay between Myofibers and Pro-Inflammatory Macrophages Controls Muscle Damage in Mdx Mice. J. Cell Sci. 2021, 134, jcs258429. [Google Scholar] [CrossRef]
- Ellwood, R.A.; Hewitt, J.E.; Torregrossa, R.; Philp, A.M.; Hardee, J.P.; Hughes, S.; van de Klashorst, D.; Gharahdaghi, N.; Anupom, T.; Slade, L.; et al. Mitochondrial Hydrogen Sulfide Supplementation Improves Health in the C. Elegans Duchenne Muscular Dystrophy Model. Proc. Natl. Acad. Sci. USA 2021, 118, e2018342118. [Google Scholar] [CrossRef]
- Orzechowska, K.; Dobrzyń, K.; Kieżun, M.; Malinowska, A.; Świderska, B.; Kamiński, T.; Smolińska, N. Chemerin Effect on the Endometrial Proteome of the Domestic Pig during Implantation Obtained by LC-MS/MS Analysis. Cells 2022, 11, 1161. [Google Scholar] [CrossRef]
- Malinowska, A.; Kistowski, M.; Bakun, M.; Rubel, T.; Tkaczyk, M.; Mierzejewska, J.; Dadlez, M. Diffprot—Software for Non-Parametric Statistical Analysis of Differential Proteomics Data. J. Proteom. 2012, 75, 4062–4073. [Google Scholar] [CrossRef]
- Elias, J.E.; Haas, W.; Faherty, B.K.; Gygi, S.P. Comparative Evaluation of Mass Spectrometry Platforms Used in Large-Scale Proteomics Investigations. Nat. Methods 2005, 2, 667–675. [Google Scholar] [CrossRef]
- Bakun, M.; Karczmarski, J.; Poznanski, J.; Rubel, T.; Rozga, M.; Malinowska, A.; Sands, D.; Hennig, E.; Oledzki, J.; Ostrowski, J.; et al. An Integrated LC-ESI-MS Platform for Quantitation of Serum Peptide Ladders. Application for Colon Carcinoma Study. Proteom. Clin. Appl. 2009, 3, 932–946. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Plubell, D.L.; Wilmarth, P.A.; Zhao, Y.; Fenton, A.M.; Minnier, J.; Reddy, A.P.; Klimek, J.; Yang, X.; David, L.L.; Pamir, N. Extended Multiplexing of Tandem Mass Tags (TMT) Labeling Reveals Age and High Fat Diet Specific Proteome Changes in Mouse Epididymal Adipose Tissue. Mol. Cell. Proteom. 2017, 16, 873–890. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Pietraszek-Gremplewicz, K.; Kozakowska, M.; Bronisz-Budzynska, I.; Ciesla, M.; Mucha, O.; Podkalicka, P.; Madej, M.; Glowniak, U.; Szade, K.; Stepniewski, J.; et al. Heme Oxygenase-1 Influences Satellite Cells and Progression of Duchenne Muscular Dystrophy in Mice. Antioxid. Redox Signal. 2018, 29, 128–148. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef]
- Le, S.; Yu, M.; Hovan, L.; Zhao, Z.; Ervasti, J.; Yan, J. Dystrophin as A Molecular Shock Absorber. ACS Nano 2018, 12, 12140–12148. [Google Scholar] [CrossRef]
- Nichols, B.; Takeda, S.; Yokota, T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains. Brain Sci. 2015, 5, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; O’Connell, J.D.; Gygi, S.P. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 2016, 27, 1620–1625. [Google Scholar] [CrossRef]
- Pigozzo, S.R.; Da Re, L.; Romualdi, C.; Mazzara, P.G.; Galletta, E.; Fletcher, S.; Wilton, S.D.; Vitiello, L. Revertant Fibers in the Mdx Murine Model of Duchenne Muscular Dystrophy: An Age- and Muscle-Related Reappraisal. PLoS ONE 2013, 8, e72147. [Google Scholar] [CrossRef]
- Arechavala-Gomeza, V.; Kinali, M.; Feng, L.; Guglieri, M.; Edge, G.; Main, M.; Hunt, D.; Lehovsky, J.; Straub, V.; Bushby, K.; et al. Revertant Fibres and Dystrophin Traces in Duchenne Muscular Dystrophy: Implication for Clinical Trials. Neuromuscul. Disord. 2010, 20, 295–301. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of Disease Specific Pathways Using in Vivo SILAC Proteomics in Dystrophin Deficient Mdx Mouse. Mol. Cell. Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef]
- Matsumura, C.Y.; Menezes de Oliveira, B.; Durbeej, M.; Marques, M.J. Isobaric Tagging-Based Quantification for Proteomic Analysis: A Comparative Study of Spared and Affected Muscles from Mdx Mice at the Early Phase of Dystrophy. PLoS ONE 2013, 8, e65831. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Characterization of Contractile Proteins from Skeletal Muscle Using Gel-Based Top-Down Proteomics. Proteomes 2019, 7, 25. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative Proteomic Analysis of the Contractile-Protein-Depleted Fraction from Normal versus Dystrophic Skeletal Muscle. Anal. Biochem. 2014, 446, 108–115. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Profiling of the Mouse Diaphragm and Refined Mass Spectrometric Analysis of the Dystrophic Phenotype. J. Muscle Res. Cell Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Multiomics Analysis of the Mdx/mTR Mouse Model of Duchenne Muscular Dystrophy. Connect. Tissue Res. 2021, 62, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Hardee, J.P.; Martins, K.J.B.; Miotto, P.M.; Ryall, J.G.; Gehrig, S.M.; Reljic, B.; Naim, T.; Chung, J.D.; Trieu, J.; Swiderski, K.; et al. Metabolic Remodeling of Dystrophic Skeletal Muscle Reveals Biological Roles for Dystrophin and Utrophin in Adaptation and Plasticity. Mol. Metab. 2021, 45, 101157. [Google Scholar] [CrossRef] [PubMed]
- Day, N.J.; Zhang, T.; Gaffrey, M.J.; Zhao, R.; Fillmore, T.L.; Moore, R.J.; Rodney, G.G.; Qian, W.-J. A Deep Redox Proteome Profiling Workflow and Its Application to Skeletal Muscle of a Duchenne Muscular Dystrophy Model. Free Radic. Biol. Med. 2022, 193, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome Analysis of the Dystrophin-Deficient MDX Diaphragm Reveals a Drastic Increase in the Heat Shock Protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef]
- Doran, P.; Dowling, P.; Lohan, J.; McDonnell, K.; Poetsch, S.; Ohlendieck, K. Subproteomics Analysis of Ca+-Binding Proteins Demonstrates Decreased Calsequestrin Expression in Dystrophic Mouse Skeletal Muscle. Eur. J. Biochem. 2004, 271, 3943–3952. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life 2022, 12, 1679. [Google Scholar] [CrossRef]
- Lee-Gannon, T.; Jiang, X.; Tassin, T.C.; Mammen, P.P.A. Biomarkers in Duchenne Muscular Dystrophy. Curr. Heart Fail. Rep. 2022, 19, 52–62. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-Free Mass Spectrometric Analysis of the Mdx-4cv Diaphragm Identifies the Matricellular Protein Periostin as a Potential Factor Involved in Dystrophinopathy-Related Fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Analysis of Dystrophin Deficiency and Associated Changes in the Aged Mdx-4cv Heart Model of Dystrophinopathy-Related Cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef]
- Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S.; Kudo, A. Periostin Is Required for the Maintenance of Muscle Fibers during Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3627. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Wakayama, Y.; Kojima, H.; Inoue, M.; Jimi, T.; Iijima, S.; Masaki, H. Aquaporin 4 Expression in the Mdx Mouse Diaphragm. Acta Histochem. Cytochem. 2011, 44, 175–182. [Google Scholar] [CrossRef]
- Liu, J.W.; Wakayama, Y.; Inoue, M.; Shibuya, S.; Kojima, H.; Jimi, T.; Oniki, H. Immunocytochemical Studies of Aquaporin 4 in the Skeletal Muscle of Mdx Mouse. J. Neurol. Sci. 1999, 164, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Frigeri, A.; Nicchia, G.P.; Nico, B.; Quondamatteo, F.; Herken, R.; Roncali, L.; Svelto, M. Aquaporin-4 Deficiency in Skeletal Muscle and Brain of Dystrophic Mdx Mice. FASEB J. 2001, 15, 90–98. [Google Scholar] [CrossRef]
- van Westering, T.L.E.; Johansson, H.J.; Hanson, B.; Coenen-Stass, A.M.L.; Lomonosova, Y.; Tanihata, J.; Motohashi, N.; Yokota, T.; Takeda, S.; Lehtiö, J.; et al. Mutation-Independent Proteomic Signatures of Pathological Progression in Murine Models of Duchenne Muscular Dystrophy. Mol. Cell. Proteom. 2020, 19, 2047–2068. [Google Scholar] [CrossRef]
- Frigeri, A.; Nicchia, G.P.; Repetto, S.; Bado, M.; Minetti, C.; Svelto, M. Altered Aquaporin-4 Expression in Human Muscular Dystrophies: A Common Feature? FASEB J. 2002, 16, 1120–1122. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence 5′-3′ |
|---|---|
| Casp3 | F: TGTCATCTCGCTCTGGTACG |
| R: AAATGACCCCTTCATCACCA | |
| Cbs | F: CCTAATTCTCACATTCTGGAC |
| R: GACACCGATGATTTTACAGC | |
| Col1a1 | F: CGATCCAGTACTCTCCGCTCTTCC |
| R: ACTACCGGGCCGATGATGCTAACG | |
| Col3a1 | F: ATCTATGAATGGTGGTTTTCA |
| R: TTTTGCAGTGGTATGTAATGT | |
| Cth | F: GAAAAGGTTGTTTATCCTGGG |
| R: CTTGATGTAGAAACTGACCATC | |
| Eef2 | F: AGAACATATTATTGCTGGCG |
| R: AACAGGGTCAGATTTCTTG | |
| Mpst | F: CGTCCTACTTGCTTTTCTC |
| R: CAGAGCTCGGAAAAGTTG | |
| Spp1 | F: CCATCTCAGAAGCAGAATCTCCTT |
| R: GGTCATGGCTTTCATTGGAATT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, O.; Myszka, M.; Podkalicka, P.; Świderska, B.; Malinowska, A.; Dulak, J.; Łoboda, A. Proteome Profiling of the Dystrophic mdx Mice Diaphragm. Biomolecules 2023, 13, 1648. https://doi.org/10.3390/biom13111648
Mucha O, Myszka M, Podkalicka P, Świderska B, Malinowska A, Dulak J, Łoboda A. Proteome Profiling of the Dystrophic mdx Mice Diaphragm. Biomolecules. 2023; 13(11):1648. https://doi.org/10.3390/biom13111648
Chicago/Turabian StyleMucha, Olga, Małgorzata Myszka, Paulina Podkalicka, Bianka Świderska, Agata Malinowska, Józef Dulak, and Agnieszka Łoboda. 2023. "Proteome Profiling of the Dystrophic mdx Mice Diaphragm" Biomolecules 13, no. 11: 1648. https://doi.org/10.3390/biom13111648
APA StyleMucha, O., Myszka, M., Podkalicka, P., Świderska, B., Malinowska, A., Dulak, J., & Łoboda, A. (2023). Proteome Profiling of the Dystrophic mdx Mice Diaphragm. Biomolecules, 13(11), 1648. https://doi.org/10.3390/biom13111648