

Actions and Consequences of Insulin in the Striatum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
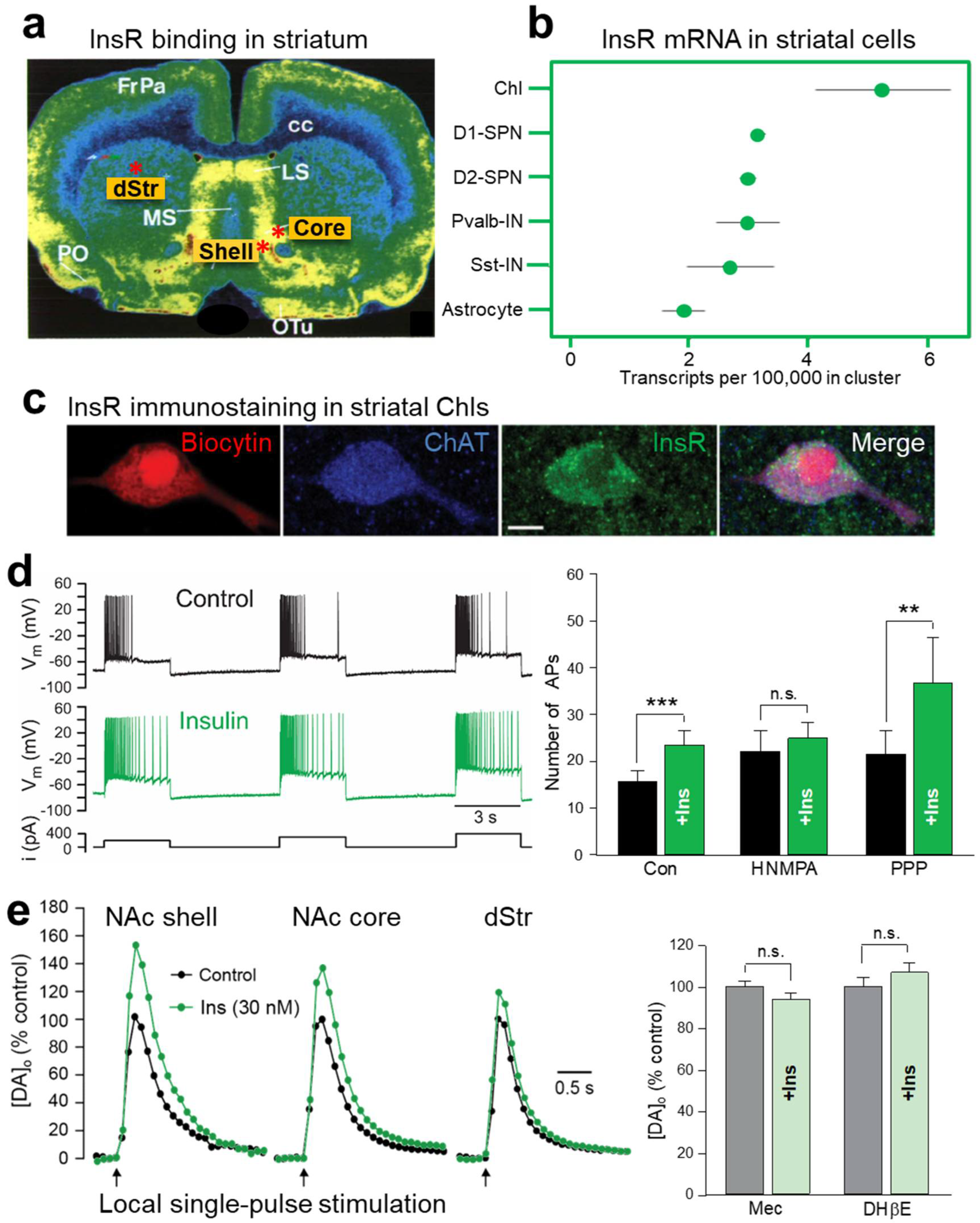
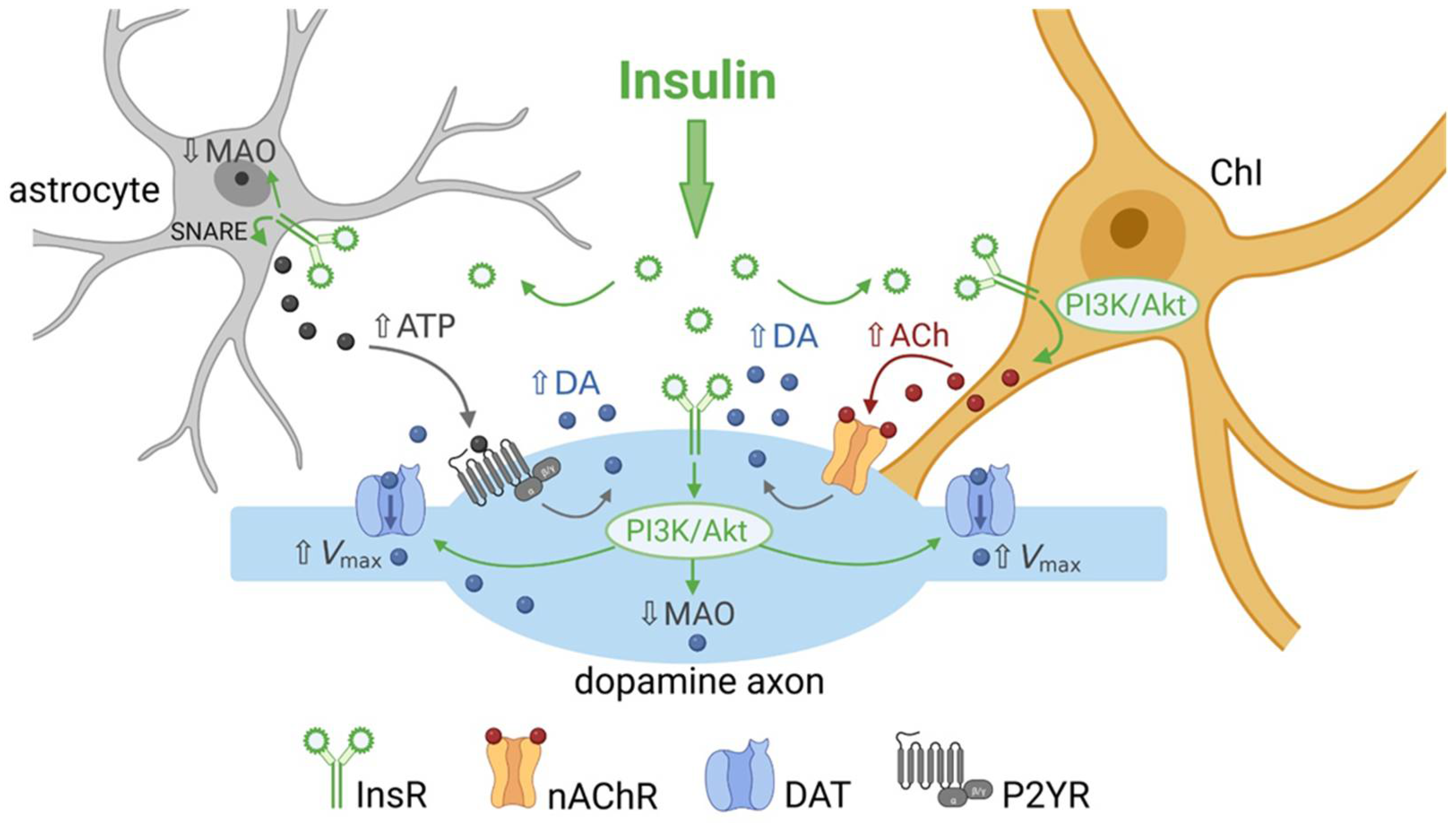
2. Insulin Promotes Striatal Dopamine Release through ChIs and nAChRs
2.1. Insulin Increases the Excitability of Striatal ChIs
2.2. Insulin Boosts Dopamine Release via InsRs, and Requires PI3K and nAChRs
3. Insulin Enhances Striatal Dopamine Uptake via DAT on Dopamine Axons
4. Insulin Inhibits Dopamine Metabolism
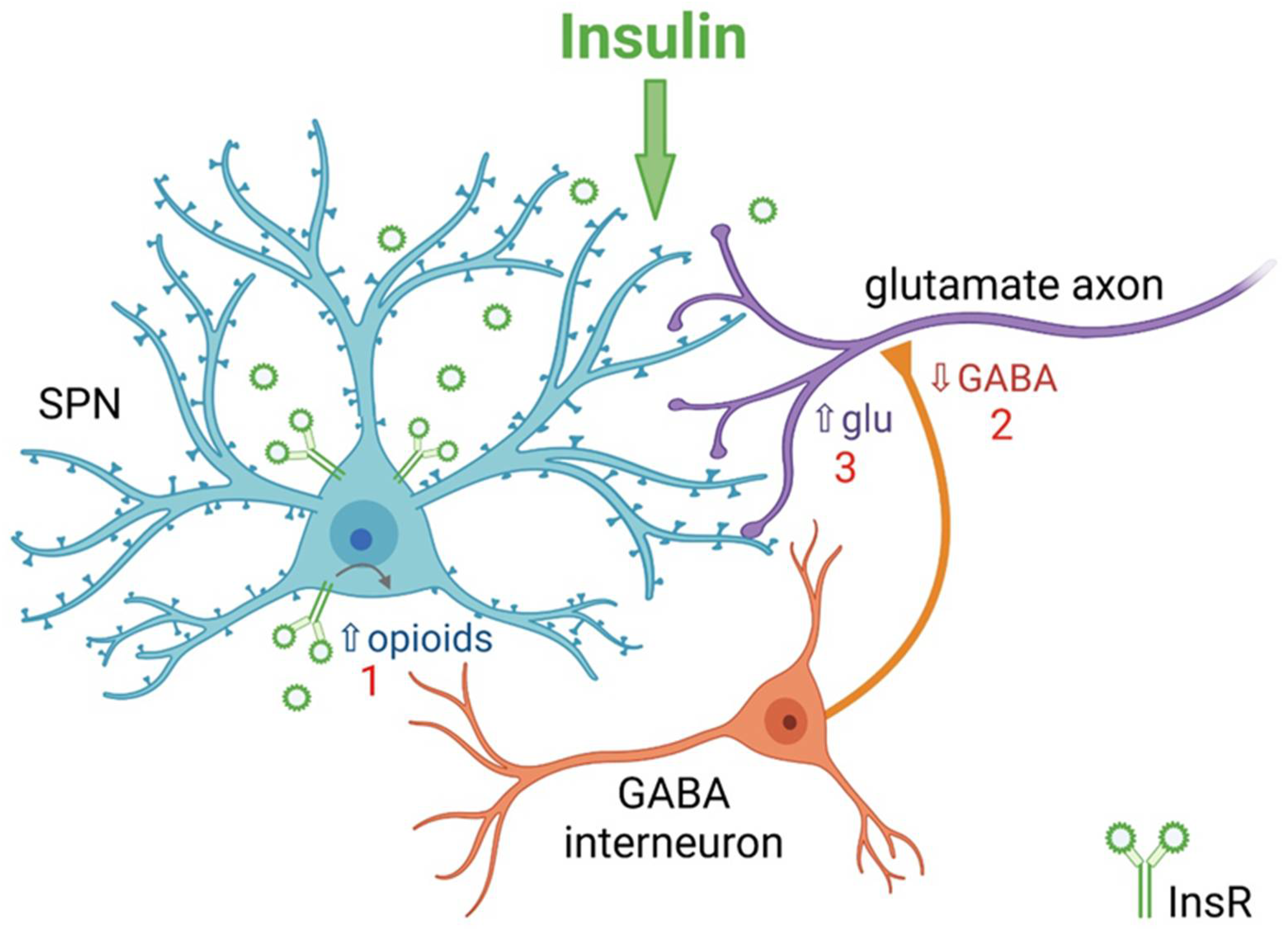
5. Insulin Bidirectionally Alters the Excitatory Regulation of Spiny Projection Neurons (SPNs)
6. Insulin Actions on Striatal Glial Cells
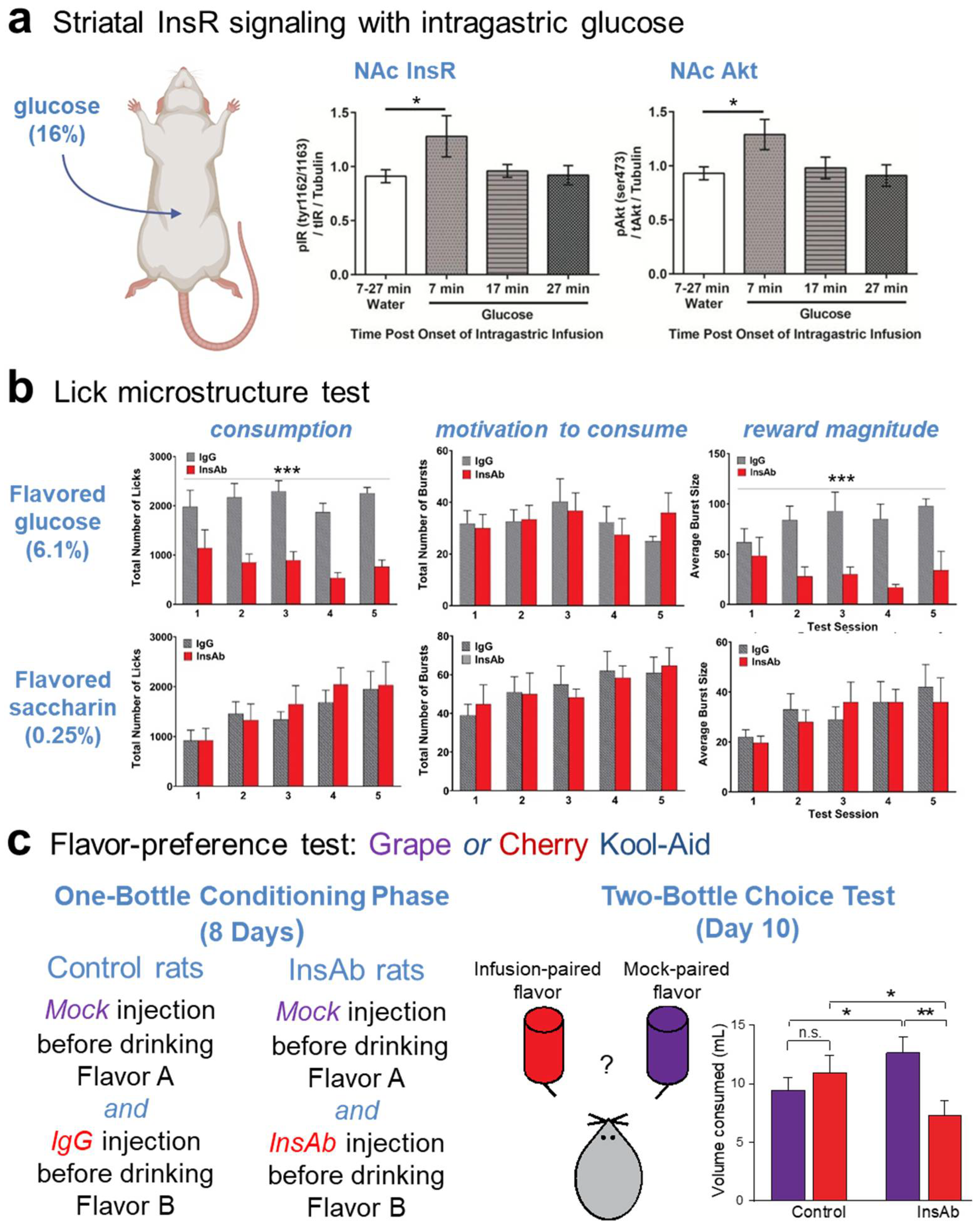
7. Role of Striatal Insulin in Signaling Food Reward and Regulating Consumption
7.1. NAc Shell Insulin Acts as a Reward Signal That Mediates Sensing of Nutrient Value
7.2. NAc Core Insulin Decreases Feeding by Impairing Motivation for Consumption
7.3. Human Imaging Studies Support a Link between Striatal Insulin and Dopamine Signaling in Food Consumption
8. Dysregulated Striatal Insulin Signaling with Diet
9. Dysregulated Striatal Insulin Signaling in Anxiety and Depression
10. Dysregulated Striatal Insulin Signaling with Age and Age-Related Disorders
11. Targeting Insulin Actions in the Striatum for Therapeutic Gain
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dagen, M.M. History of Insulin. In Encyclopedia of Life Sciences (eLS); John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Barrett, E.J. Insulin transport into the brain. Am. J. Physiol. Cell. Physiol. 2018, 315, C125–C136. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y. Origins of brain insulin and its function. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; pp. 1–11. [Google Scholar]
- Mazucanti, C.H.; Liu, Q.R.; Lang, D.; Huang, N.; O’Connell, J.F.; Camandola, S.; Egan, J.M. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight 2019, 4, e131682. [Google Scholar] [CrossRef] [PubMed]
- Molnár, G.; Faragó, N.; Kocsis, Á.K.; Rózsa, M.; Lovas, S.; Boldog, E.; Báldi, R.; Csajbók, É.; Gardi, J.; Puskás, L.G.; et al. GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J. Neurosci. 2014, 34, 1133–1137. [Google Scholar] [CrossRef]
- Csajbók, É.A.; Kocsis, Á.K.; Faragó, N.; Furdan, S.; Kovács, B.; Lovas, S.; Molnár, G.; Likó, I.; Zvara, Á.; Puskás, L.G.; et al. Expression of GLP-1 receptors in insulin-containing interneurons of rat cerebral cortex. Diabetologia 2019, 62, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Aβ oligomers. Mol. Biol. Cell 2017, 28, 2623–2636. [Google Scholar] [CrossRef]
- Strubbe, J.H.; Porte, D., Jr.; Woods, S.C. Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in the rat. Physiol. Behav. 1988, 44, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J. Differential permeability of the blood-brain barrier to two pancreatic peptides: Insulin and amylin. Peptides 1998, 19, 883–889. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef]
- Woods, S.C.; Seeley, R.J.; Baskin, D.G.; Schwartz, M.W. Insulin and the blood brain barrier. Curr. Pharm. Des. 2003, 9, 795–800. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic localization of insulin receptors in rat brain: Prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570. [Google Scholar] [CrossRef]
- Schulingkamp, R.J.; Pagano, T.C.; Hung, D.; Raffa, R.B. Insulin receptors and insulin action in the brain: Review and clinical implications, Neurosci. Biobehav. Rev. 2000, 24, 855–872. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Evans, S.B.; Murphy, J.; Hoen, M.; Baskin, D.G. Expression of receptors for insulin and leptin in ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003, 964, 107–115. [Google Scholar] [CrossRef]
- Stouffer, M.A.; Woods, C.A.; Patel, J.C.; Lee, C.R.; Witkovsky, P.; Bao, L.; Machold, R.P.; Jones, K.T.; de Vaca, S.C.; Reith, M.E.; et al. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun. 2015, 6, 8543. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat: Historical perspective. Brain Res. 2016, 1645, 68–70. [Google Scholar] [CrossRef]
- Bondy, C.A.; Cheng, C.M. Signaling by insulin-like growth factor 1 in brain. Eur. J. Pharmacol. 2004, 490, 25–31. [Google Scholar] [CrossRef]
- Vigneri, R.; Squatrito, S.; Sciacca, L. Insulin and its analogs: Actions via insulin and IGF receptors. Acta. Diabetol. 2010, 47, 271–278. [Google Scholar] [CrossRef]
- Bevan, P. Insulin signalling. J. Cell Sci. 2001, 114, 1429–1430. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Tseng, Y.H.; Kahn, C.R. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J. Biol. Chem. 2010, 285, 17235–17245. [Google Scholar] [CrossRef]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Bellisle, F.; Drewnowski, A.; Anderson, G.H.; Westerterp-Plantenga, M.; Martin, C.K. Sweetness, satiation, and satiety. J. Nutr. 2012, 142, 1149S–1154S. [Google Scholar] [CrossRef]
- Tiedemann, L.J.; Schmid, S.M.; Hettel, J.; Giesen, K.; Francke, P.; Büchel, C.; Brassen, S. Central insulin modulates food valuation via mesolimbic pathways. Nat. Commun. 2017, 8, 16052. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Reagan, L.P. Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology 2018, 136, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Pothos, E.N. Impact of brain insulin signaling on dopamine function, food intake, reward, and emotional behavior. Curr. Nutr. Rep. 2019, 8, 83–91. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Häring, H.U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Sallam, N.A.; Borgland, S.L. Insulin and endocannabinoids in the mesolimbic system. J. Neuroendocrinol. 2021, 33, e12965. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cai, W.; Hoover, B.; Kahn, C.R. Insulin action in the brain: Cell types, circuits and diseases. Trends Neurosci. 2022, 45, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.R.; Finnell, J.E. Beyond the hypothalamus: Roles for insulin as a regulator of neurotransmission, motivation, and feeding. Neuropsychopharmacology 2023, 48, 232–233. [Google Scholar] [CrossRef]
- Mebel, D.M.; Wong, J.C.; Dong, Y.J.; Borgland, S.L. Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur. J. Neurosci. 2012, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Labouébe, G.; Liu, S.; Dias, C.; Zou, H.; Wong, J.C.; Karunakaran, S.; Clee, S.M.; Phillips, A.G.; Boutrel, B.; Borgland, S.L. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci. 2013, 16, 300–308. [Google Scholar] [CrossRef]
- Naef, L.; Seabrook, L.; Hsiao, J.; Li, C.; Borgland, S.L. Insulin in the ventral tegmental area reduces cocaine-evoked dopamine in the nucleus accumbens in vivo. Eur. J. Neurosci. 2019, 50, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Cachope, R.; Mateo, Y.; Mathur, B.N.; Irving, J.; Wang, H.L.; Morales, M.; Lovinger, D.M.; Cheer, J.F. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: Setting the tone for reward processing. Cell Rep. 2012, 2, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; Lalic, T.; Platt, N.J.; Jennings, K.A.; Deisseroth, K.; Cragg, S.J. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 2012, 75, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cai, X.; Ritzau-Jost, A.; Kramer, P.; Li, Y.; Khaliq, Z.M.; Hallermann, S.; Kaeser, P.S. An action potential intiation mechanism in distal axons for the control of dopamine release. Science 2022, 375, 1378–1385. [Google Scholar] [CrossRef]
- Kramer, P.F.; Brill-Weil, S.G.; Cummins, A.C.; Zhang, R.; Camacho-Hernandez, G.A.; Newman, A.H.; Eldridge, M.A.G.; Averbeck, B.B.; Khaliq, Z.M. Synaptic-like axo-axonal transmission from striatal cholinergic interneurons onto dopaminergic fibers. Neuron 2022, 18, 2949–2960. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bein, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef]
- Sanchez, G.; Rodriguez, M.J.; Pomata, P.; Rela, L.; Murer, M.G. Reduction of an afterhyperpolarization current increases excitability in striatal cholinergic interneurons in rat parkinsonism. J. Neurosci. 2011, 31, 6553–6564. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Callaway, J.C.; Wilson, C.J. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J. Neurosci. 2000, 20, 8493–8503. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.C.; Rice, M.E. Monitoring axonal and somatodendritic dopamine release using fast-scan cyclic voltammetry in brain slices. Methods Mol. Biol. 2013, 96, 243–273. [Google Scholar]
- Patel, J.C. Voltammetry: Electrochemical detection of neurotransmitters in the brain. In Encyclopedia of Life Sciences (eLS); John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar]
- Rice, M.E.; Cragg, S.J. Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 2004, 7, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.C.; Rossignol, E.; Rice, M.E.; Machold, R.P. Opposing regulation of striatal dopamine release and exploratory motor behavior by forebrain and brainstem cholinergic inputs. Nat. Commun. 2012, 3, 1172. [Google Scholar] [CrossRef]
- Rice, M.E.; Patel, J.C.; Cragg, S.J. Dopamine release in the basal ganglia. Neuroscience 2011, 198, 112–137. [Google Scholar] [CrossRef]
- Sulzer, D.; Cragg, S.J.; Rice, M.E. Striatal dopamine neurotransmission: Regulation of release and uptake. Basal Ganglia 2016, 6, 123–148. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK378978/ (accessed on 24 May 2022).
- Nirenberg, M.J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J. Neurosci. 1996, 16, 436–447. [Google Scholar] [CrossRef]
- Nirenberg, M.J.; Chan, J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. Immunogold localization of the dopamine transporter: An ultrastructural study of the rat ventral tegmental area. J. Neurosci. 1997, 17, 5255–5262. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Yi, H.; Heilman, C.J.; Edwards, R.H.; Levey, A.I. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra. J. Comp. Neurol. 1997, 388, 211–227. [Google Scholar] [CrossRef]
- Bu, M.; Farrer, M.J.; Khoshbouei, H. Dynamic control of the dopamine transporter in neurotransmission and homeostasis. NPJ Parkinsons Dis. 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Ingram, S.L.; Scimemi, A. Regulation of Glutamate, GABA and Dopamine Transporter Uptake, Surface Mobility and Expression. Front. Cell. Neurosci. 2021, 15, 670346. [Google Scholar] [CrossRef]
- Jones, S.R.; Gainetdinov, R.R.; Jaber, M.; Giros, B.; Wightman, R.M.; Caron, M.G. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc. Natl. Acad. Sci. USA 1998, 95, 4029–4034. [Google Scholar] [CrossRef]
- Figlewicz, D.P.; Szot, P.; Chavez, M.; Woods, S.C.; Veith, R.C. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994, 644, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, L.; Moron, J.A.; Kahlig, K.M.; Ferrer, J.V.; Sen, N.; Lechleiter, J.D.; Leeb-Lundberg, L.M.F.; Merrill, G.; Lafer, E.M.; Ballou, L.M.; et al. PI3-kinase regulation of dopamine uptake. J. Neurochem. 2002, 81, 859–869. [Google Scholar] [CrossRef]
- Garcia, B.G.; Wei, Y.; Moron, J.A.; Lin, R.Z.; Javitch, J.A.; Galli, A. Akt is essential for insulin modulation of amphetamine-induced human dopamine transporter cell-surface redistribution. Mol. Pharmacol. 2005, 68, 102–109. [Google Scholar] [CrossRef]
- Fagan, R.R.; Kearney, P.J.; Melikian, H.E. In Situ regulated dopamine transporter trafficking: There’s no place like home. Neurochem. Res. 2020, 45, 1335–1343. [Google Scholar] [CrossRef]
- Schoffelmeer, A.N.; Drukarch, B.; De Vries, T.J.; Hoenboom, F.; Schetters, D.; Pattij, T. Insulin modulates cocaine-sensitive monoamine transporter function and impulsive behavior. J. Neurosci. 2011, 31, 1284–1291. [Google Scholar] [CrossRef]
- Jones, K.T.; Woods, C.; Zhen, J.; Antonio, T.; Carr, K.D.; Reith, M.E.A. Effects of diet and insulin on dopamine-transporter activity and expression in rat caudate-putamen, nucleus accumbens, and midbrain. J. Neurochem. 2017, 140, 728–740. [Google Scholar] [CrossRef]
- Patel, J.C.; Stouffer, M.A.; Mancini, M.; Nicholson, C.; Carr, K.D.; Rice, M.E. Interactions between insulin and diet on striatal dopamine uptake kinetics in rodent brain slices. Eur. J. Neurosci. 2019, 49, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Wightman, R.M.; Amatore, C.; Engstrom, R.C.; Hale, P.D.; Kristensen, E.W.; Kuhr, W.G.; May, L.J. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience 1988, 25, 513–523. [Google Scholar] [CrossRef]
- Nicholson, C. Interaction between diffusion and Michaelis-Menten uptake of dopamine after iontophoresis in striatum. Biophys. J. 1995, 68, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Fordahl, S.C.; Jones, S.R. High-fat-diet-induced deficits in dopamine terminal function are reversed by restoring insulin signaling. ACS Chem. Neurosci. 2017, 8, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.F.; O’Dell, S.J.; Navarrete, R.; Rosenstein, A.J. Dopamine high-affinity transport site topography in rat brain: Major differences between dorsal and ventral striatum. Neuroscience 1990, 37, 11–21. [Google Scholar] [CrossRef]
- Ciliax, B.J.; Heilman, C.; Demchyshyn, L.L.; Pristupa, Z.B.; Ince, E.; Hersch, S.M.; Niznik, H.B.; Levey, A.I. The dopamine transporter: Immunochemical characterization and localization in brain. J. Neurosci. 1995, 15, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, T.; Barroso-Chinea, P.; De La Cruz Muros, I.; Del Mar Perez-Delgado, M.; Rodriguez, M. Expression of dopamine and vesicular monoamine transporters and differential vulnerability of mesostriatal dopaminergic neurons. J. Comp. Neurol. 2004, 479, 198–215. [Google Scholar] [CrossRef]
- Condon, M.D.; Platt, N.J.; Zhang, Y.F.; Roberts, B.M.; Clements, M.A.; Vietti-Michelina, S.; Tseu, M.Y.; Brimblecombe, K.R.; Threlfell, S.; Mann, E.O.; et al. Plasticity in striatal dopamine release is governed by release-independent depression and the dopamine transporter. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Orosco, M.; Rouch, C.; Gripois, D.; Blouquit, M.F.; Roffi, J.; Jacquot, C.; Cohen, Y. Striatal dopamine metabolism is differentially affected by insulin according to the genotype in Zucker rats: A microdialysis study. Psychoneuroendocrinology 1992, 17, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R.; Surmeier, D.J. Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef]
- Fetterly, T.L.; Oginsky, M.F.; Nieto, A.M.; Alonso-Caraballo, Y.; Santana-Rodriguez, Z.; Ferrario, C.R. Insulin bidirectionally alters NAc glutamatergic transmission: Interactions between insulin receptor activation, endogenous opioids, and glutamate release. J. Neurosci. 2021, 41, 2360–2372. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Hasel, P.; Liddelow, S.A. Astrocytes. Curr. Biol. 2021, 31, R326–R327. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Parpura, V.; Li, B.; Scuderi, C. Astrocytes: The housekeepers and guardians of the CNS. Adv. Neurobiol. 2021, 26, 21–53. [Google Scholar] [PubMed]
- Goubard, V.; Fino, E.; Venance, L. Contribution of astrocytic glutamate and GABA uptake to corticostriatal information processing. J. Physiol. 2011, 589, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter release from astrocytes: Functional, developmental, and pathological implications in the brain. Front. Neurosci. 2015, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic cells in the brain. Front. Cell. Neurosci. 2022, 16, 892497. [Google Scholar] [CrossRef]
- Sa, M.; Park, M.G.; Lee, C.J. Role of hypothalamic reactive astrocytes in diet-induced obesity. Mol. Cell 2022, 45, 65–75. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin action on astrocytes: From energy homeostasis to behaviour. J. Neuroendocrinol. 2021, 33, e12953. [Google Scholar] [CrossRef]
- Heni, M.; Hennige, A.M.; Peter, A.; Siegel-Axel, D.; Ordelheide, A.M.; Krebs, N.; Machicao, F.; Fritsche, A.; Häring, H.U.; Staiger, H. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS ONE 2011, 6, e21594. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef] [PubMed]
- Roitman, M.F.; Stuber, G.D.; Phillips, P.E.M.; Wightman, R.M.; Carelli, R.M. Dopamine operates as a subsecond modulator of food seeking. J. Neurosci. 2004, 24, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007, 30, 375–381. [Google Scholar] [CrossRef]
- Zhou, Q.Y.; Palmiter, R.D. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 1995, 83, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, I.E. Circuit organization of sugar reinforcement. Physiol. Behav. 2016, 164, 473–477. [Google Scholar] [CrossRef]
- Rossi, M.A.; Stuber, G.D. Overlapping brain circuits for homeostatic and hedonic feeding. Cell Metab. 2018, 27, 42–56. [Google Scholar] [CrossRef]
- Kullmann, S.; Blum, D.; Jaghutriz, B.A.; Gassenmaier, C.; Bender, B.; Häring, H.U.; Reischl, G.; Preissl, H.; la Fougère, C.; Fritsche, A.; et al. Central insulin modulates dopamine signaling in the human striatum. J. Clin. Endocrinol. Metab. 2021, 106, 2949–2961. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J. Reward mechanisms in obesity: New insights and future directions. Neuron 2011, 69, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.R.; Münzberg, H.; Morrison, C.D. Blaming the brain for obesity: Integration of hedonic and homeostatic mechanisms. Gastroenterology 2017, 152, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; McCutcheon, J.E.; Roitman, M.F. Parallels and overlap: The integration of homeostatic signals by mesolimbic dopamine neurons. Front. Psychiatry 2018, 9, 410. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.D. Modulatory effects of food restriction on brain and behavioral effects of abused drugs. Curr. Pharm. Des. 2020, 26, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef]
- Woods, C.A.; Guttman, Z.R.; Huang, D.; Kolaric, R.A.; Rabinowitsch, A.I.; Jones, K.T.; Cabeza de Vaca, S.; Sclafani, A.; Carr, K.D. Insulin receptor activation in the nucleus accumbens reflects nutritive value of a recently ingested meal. Physiol. Behav. 2016, 159, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.D.; Weiner, S.P. Effects of nucleus accumbens insulin inactivation on microstructure of licking for glucose and saccharin in male and female rats. Physiol. Behav. 2022, 249, 113769. [Google Scholar] [CrossRef]
- Finnell, J.E.; Ferrario, C.R. Intra-NAc insulin reduces the motivation for food and food intake without altering cue-triggered food-seeking. Physiol. Behav. 2022, 254, 113892. [Google Scholar] [CrossRef]
- Davis, J.D.; Smith, G.P. Analysis of the microstructure of the rhythmic tongue movements of rats ingesting maltose and sucrose solutions. Behav. Neurosci. 1992, 106, 217–228. [Google Scholar] [CrossRef]
- Spector, A.C.; Klumpp, P.A.; Kaplan, J.M. Analytical issues in the evaluation of food deprivation and sucrose concentration effects on the microstructure of licking behavior in the rat. Behav. Neurosci. 1998, 112, 678–694. [Google Scholar] [CrossRef]
- Lardeaux, S.; Kim, J.J.; Nicola, S.M. Intermittent access to sweet high-fat liquid induces increased palatability and motivation to consume in a rat model of binge consumption. Physiol. Behav. 2013, 10, 114–115. [Google Scholar]
- Naneix, F.; Peters, K.Z.; McCutcheon, J.E. Investigating the effect of physiological need states on palatability and motivation using microstructural analysis of licking. Neuroscience 2020, 447, 155–166. [Google Scholar] [CrossRef]
- Davis, J.D.; Smith, G.P.; Kung, T.M. Cholecystokinin changes the duration but not the rate of licking in vagotomized rats. Behav. Neurosci. 1995, 109, 991–996. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Donahey, J.; Baird, J.-P.; Simansky, K.J.; Grill, H.J. d-Fenfluramine anorexia: Dissociation of ingestion rate, meal duration, and meal size effects. Pharmacol. Biochem. Behav. 1997, 57, 223–229. [Google Scholar] [CrossRef]
- Eisen, S.; Davis, J.D.; Rauhofer, E.; Smith, G.P. Gastric negative feedback produced by volume and nutrient during a meal in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R1201–R1214. [Google Scholar] [CrossRef]
- Hsiao, S.; Fan, R.J. Additivity of taste-specific effects of sucrose and quinine: Microstructural analysis of ingestive behavior in rats. Behav. Neurosci. 1993, 107, 317–326. [Google Scholar] [CrossRef]
- Dwyer, D.M.; Lydall, E.S.; Hayward, A.J. Simultaneous contrast: Evidence from licking microstructure and cross-solution comparisons. J. Exp. Psychol. Anim. Behav. Process. 2011, 37, 200–210. [Google Scholar] [CrossRef]
- McCutcheon, J.E. The role of dopamine in the pursuit of nutritional value. Physiol. Behav. 2015, 152, 408–415. [Google Scholar] [CrossRef]
- Genn, R.F.; Higgs, C.; Cooper, S.J. The effects of 7-OH-DPAT, quinpirole and raclopride on licking for sucrose solutions in the non-deprived rat. Behav. Pharmacol. 2003, 14, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.H.; Davis, J.D.; Watson, C.A.; Smith, G.P. Similar effect of raclopride and reduced sucrose concentration on the microstructure of sucrose sham feeding, Eur. J. Pharmacol. 1990, 186, 61–70. [Google Scholar]
- Touzani, K.; Bodnar, R.; Sclafani, A. Activation of dopamine D1-like receptors in nucleus accumbens is critical for the acquisition, but not the expression, of nutrient conditioned flavor preferences in rats. Eur. J. Neurosci. 2008, 27, 1525–1533. [Google Scholar] [CrossRef]
- Sclafani, A.; Touzani, K.; Bodnar, R.J. Dopamine and learned food preferences. Physiol. Behav. 2011, 104, 64–68. [Google Scholar] [CrossRef]
- Mark, G.P.; Rada, P.; Pothos, E.; Hoebel, B.G. Effects of feeding and drinking on acetylcholine release in the nucleus accumbens, striatum, and hippocampus of freely behaving rats. J. Neurochem. 1992, 58, 2269–2274. [Google Scholar] [CrossRef]
- Avena, N.M.; Rada, P.; Moise, N.; Hoebel, B.G. Sucrose sham feeding on a binge schedule releases accumbens dopamine repeatedly and eliminates the acetylcholine satiety response. Neuroscience 2006, 139, 813–820. [Google Scholar] [CrossRef]
- Reichenbach, A.; Clarke, R.E.; Stark, R.; Lockie, S.H.; Mequinion, M.; Dempsey, H.; Rawlinson, S.; Reed, F.; Sepehrizadeh, T.; DeVeer, M.; et al. Metabolic sensing in AgRP neurons integrates homeostatic state with dopamine signalling in the striatum. Elife 2022, 11, e72668. [Google Scholar] [CrossRef]
- McCaleb, M.L.; Myers, R.D. Striatal dopamine release is altered by glucose and insulin during push-pull perfusion of the rat’s caudate nucleus. Brain Res. Bull. 1979, 4, 651–656. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Jeanrenaud, B. Sham feeding-induced cephalic phase insulin release in the rat. Am. J. Physiol. 1982, 242, E280–E285. [Google Scholar] [CrossRef]
- Thanarajah, S.E.; Backes, H.; DiFeliceantonio, A.G.; Albus, K.; Cremer, A.L.; Hanssen, R.; Lippert, R.N.; Cornely, O.A.; Small, D.M.; Brüning, J.C.; et al. Food intake recruits orosensory and post-ingestive dopaminergic circuits to affect eating desire in humans. Cell Metab. 2019, 29, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Jones-Gotman, M.; Dagher, A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage 2003, 19, 1709–1715. [Google Scholar] [CrossRef]
- Tellez, L.A.; Han, W.; Zhang, X.; Ferreira, T.L.; Perez, I.O.; Shammah-Lagnado, S.J.; van den Pol, A.N.; de Araujo, I.E. Separate circuitries encode the hedonic and nutritional values of sugar. Nat. Neurosci. 2016, 19, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.D. Chronic food restriction: Enhancing effects on drug reward and striatal cell signaling. Physiol. Behav. 2007, 91, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.D. Food scarcity, neuroadaptations, and the pathogenic potential of dieting in an unnatural ecology: Binge eating and drug abuse. Physiol. Behav. 2011, 104, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.F.; Tracy, A.L.; Schurdak, J.D.; Tsch€op, M.H.; Lipton, J.W.; Clegg, D.J.; Benoit, S.C. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav. Neurosci. 2008, 122, 1257–1263. [Google Scholar] [CrossRef]
- Stice, E.; Figlewicz, D.P.; Gosnell, B.A.; Levine, A.S.; Pratt, W.E. The contribution of brain reward circuits to the obesity epidemic. Neurosci. Biobehav. Rev. 2013, 37, 2047–2058. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Labouébe, G.; Liu, S.; Nieh, E.H.; Routh, V.H.; Xu, S.; O’Connor, E.C. Homeostasis meets motivation in the battle to control food intake. J. Neurosci. 2016, 36, 11469–11481. [Google Scholar] [CrossRef]
- Ferrario, C.R. Why did I eat that? Contributions of individual differences in incentive motivation and nucleus accumbens plasticity to obesity. Physiol. Behav. 2020, 227, 113114. [Google Scholar]
- Gnazzo, F.G.; Mourra, D.; Guevara, C.A.; Beeler, J.A. Chronic food restriction enhances dopamine-mediated intracranial self-stimulation. Neuroreport 2021, 32, 1128–1133. [Google Scholar] [CrossRef]
- Kullmann, S.; Valenta, V.; Wagner, R.; Tschritter, O.; Machann, J.; Häring, H.U.; Preissl, H.; Fritsche, A.; Heni, M. Brain insulin sensitivity is linked to adiposity and body fat distribution. Nat. Commun. 2020, 11, 1841. [Google Scholar] [CrossRef]
- Heni, M.; Wagner, R.; Kullmann, S.; Gancheva, S.; Roden, M.; Peter, A.; Stefan, N.; Preissl, H.; Häring, H.-U.; Fritsche, A. Hypothalamic and striatal insulin action suppresses endogenous glucose production and may stimulate glucose uptake during hyperinsulinemia in lean but not in overweight men. Diabetes 2017, 66, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.C.; Kumar, N.S.; Inglis, M.A.; Anderson, G.M. Leptin and insulin do not exert redundant control of metabolic or emotive function via dopamine neurons. Horm. Behav. 2018, 106, 93–104. [Google Scholar] [PubMed]
- Tchilian, E.Z.; Zhelezarov, I.E.; Petkov, V.V.; Hadjiivanova, C.I. 125I-insulin binding is decreased in olfactory bulbs of aged rats. Neuropeptides 1990, 17, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Kar, S.; Rowe, W.; Quirion, R. Distribution and levels of [125I]IGF-I, [125I]IGF-II and [125I]insulin receptor binding sites in the hippocampus of aged memory-unimpaired and -impaired rats. Neuroscience 1997, 80, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Zaia, A.; Piantanelli, L. Insulin receptors in the brain cortex of aging mice. Mech. Ageing Dev. 2000, 113, 227–232. [Google Scholar] [CrossRef]
- Pijpers, E.; Ferreira, I.; de Jongh, R.T.; Deeg, D.J.; Lips, P.; Stehouwer, C.D.; Nieuwenhuijzen Kruseman, A.C. Older individuals with diabetes have an increased risk of recurrent falls: Analysis of potential mediating factors: The longitudinal ageing study Amsterdam. Age Ageing 2012, 41, 358–365. [Google Scholar]
- Crews, R.T.; Yalla, S.V.; Fleischer, A.E.; Wu, S.C. A growing troubling triad: Diabetes, aging, and falls. J. Aging Res. 2013, 2013, 342650. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, X.; Zhang, Q.; Zou, R. Diabetes mellitus and risk of falls in older adults: A systematic review and meta-analysis. Age Ageing 2016, 45, 761–767. [Google Scholar] [CrossRef]
- Case, S.L.; Frazier, H.N.; Anderson, K.L.; Lin, R.-L.; Thibault, O. Falling Short: The contribution of central insulin receptors to gait dysregulation in brain aging. Biomedicines 2022, 10, 1923. [Google Scholar] [CrossRef]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and insulin resistance in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef] [PubMed]
- Fiory, F.; Perruolo, G.; Cimmino, I.; Cabaro, S.; Pignalosa, F.C.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. The relevance of insulin action in the dopaminergic system. Front. Neurosci. 2019, 13, 868. [Google Scholar] [CrossRef]
- Yu, H.; Sun, T.; He, X.; Wang, Z.; Zhao, K.; An, J.; Wen, L.; Li, J.Y.; Li, W.; Feng, J. Association between Parkinson’s disease and diabetes mellitus: From epidemiology, pathophysiology and prevention to treatment. Aging Dis. 2022, 13, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The association between type 2 diabetes mellitus and Parkinson’s disease. J. Parkinsons Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Komici, K.; Femminella, G.D.; Bencivenga, L.; Rengo, G.; Pagano, G. Diabetes mellitus and Parkinson’s disease: A systematic review and meta-analyses. J. Parkinsons Dis. 2021, 11, 1585–1596. [Google Scholar] [CrossRef]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [PubMed]
- Wang, L.; Zhai, Y.Q.; Xu, L.L.; Qiao, C.; Sun, X.L.; Ding, J.H.; Ming, L.; Gang, H. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp. Neurol. 2014, 251, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Perruolo, G.; Viggiano, D.; Fiory, F.; Cassese, A.; Nigro, C.; Liotti, A.; Miele, C.; Beguinot, F.; Formisano, P. Parkinson-like phenotype in insulin-resistant PED/PEA-15 transgenic mice. Sci. Rep. 2016, 6, 29967. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef]
- Morris, J.K.; Zhang, H.; Gupte, A.A.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Measures of striatal insulin resistance in a 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2008, 1240, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The role of Insulin/IGF-1/PI3K/Akt/GSK3β Signaling in Parkinson’s disease dementia. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef]
- Bäckman, L.; Ginovart, N.; Dixon, R.A.; Wahlin, T.B.; Wahlin, A.; Halldin, C.; Farde, L. Age-related cognitive deficits mediated by changes in the striatal dopamine system. Am. J. Psychiatry 2000, 157, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Pignalosa, F.C.; Desiderio, A.; Mirra, P.; Nigro, C.; Perruolo, G.; Ulianich, L.; Formisano, P.; Beguinot, F.; Miele, C.; Napoli, R.; et al. Diabetes and cognitive impairment: A role for glucotoxicity and dopaminergic dysfunction. Int. J. Mol. Sci. 2021, 22, 12366. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de La Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease: Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed]
- DeFelice, F.G.; Gonçalves, R.A.; Ferreira, S.T. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat. Rev. Neurosci. 2022, 23, 215–230. [Google Scholar] [CrossRef]
- Speed, N.; Saunders, C.; Davis, A.R.; Owens, W.A.; Matthies, H.J.G.; Saadat, S.; Kennedy, J.P.; Vaughan, R.A.; Neve, R.L.; Lindsley, C.W.; et al. Impaired striatal Akt signaling disrupts dopamine homeostasis and increases feeding. PLoS ONE 2011, 6, e25169. [Google Scholar] [CrossRef]
- Mancini, M.; Patel, J.C.; Affinati, A.H.; Witkovsky, P.; Rice, M.E. Leptin promotes striatal dopamine release via cholinergic interneurons and regionally distinct signaling pathways. J. Neurosci. 2022, 42, 6668–6679. [Google Scholar] [CrossRef]
- Luppino, F.S.; deWit, L.M.; Bouvy, P.F.; Stijnen, T.; Cuijpers, P.; Penninx, B.W.; Zitman, F.G. Overweight, obesity, and depression: A systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 2010, 67, 220–229. [Google Scholar] [CrossRef]
- Eaton, W.W.; Armenian, H.; Gallo, J.; Pratt, L.; Ford, D.E. Depression and risk for onset of type II diabetes. A prospective population-based study. Diabetes Care 1996, 19, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Jantaratnotai, N.; Mosikanon, K.; Lee, Y.; McIntyre, R.S. The interface of depression and obesity. Obes. Res. Clin. Pract. 2017, 11, 1–10. [Google Scholar] [CrossRef]
- Sevilla-González, M.D.R.; Quintana-Mendoza, B.M.; Aguilar-Salinas, C.A. Interaction between depression, obesity, and type 2 diabetes: A complex picture. Arch. Med. Res. 2017, 48, 582–591. [Google Scholar] [CrossRef]
- Borgland, S.L. Can treatment of obesity reduce depression or vice versa? J. Psychiatry Neurosci. 2021, 46, E313–E318. [Google Scholar] [CrossRef]
- Bastioli, G.; Arnold, J.C.; Mancini, M.; Mar, A.C.; Gamallo-Lana, B.; Saadipour, K.; Chao, M.V.; Rice, M.E. Voluntary exercise boosts striatal dopamine release: Evidence for the necessary and sufficient role of BDNF. J. Neurosci. 2022, 42, 4725–4736. [Google Scholar] [PubMed]
- Chen, W.; Li, J.; Liu, J.; Wang, D.; Hou, L. Aerobic exercise improves food reward systems in obese rats via insulin signaling regulation of dopamine levels in the nucleus accumbens. ACS Chem. Neurosci. 2019, 10, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.L.; Lee, M.S.; Hsu, C.C.; Chuang, S.Y.; Lee, J.T.; Tsai, H.N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012, 18, 753–758. [Google Scholar] [PubMed]
- Cardoso, S.; Moreira, P.I. Antidiabetic drugs for Alzheimer’s and Parkinson’s diseases: Repurposing insulin, metformin, and thiazolidinediones. Int. Rev. Neurobiol. 2020, 155, 37–64. [Google Scholar] [PubMed]
- Nowell, J.; Blunt, E.; Edison, P. Incretin and insulin signaling as novel therapeutic targets for Alzheimer’s and Parkinson’s disease. Mol. Psych. 2023, 28, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [PubMed]
- Benedict, C.; Hallschmid, M.; Schultes, B.; Born, J.; Kern, W. Intranasal insulin to improve memory function in humans. Neuroendocrinology 2007, 86, 136–142. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal insulin. J. Neuroendocrinol. 2021, 33, e12934. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Benedict, C.; Born, J.; Kern, W. Targeting metabolic and cognitive pathways of the CNS by intranasal insulin administration. Expert Opin. Drug Deliv. 2007, 4, 319–322. [Google Scholar] [CrossRef]
- Pang, Y.; Lin, S.; Wright, C.; Shen, J.; Carter, K.; Bhatt, A.; Fan, L.W. Intranasal insulin protects against substantia nigra dopaminergic neuronal loss and alleviates motor deficits induced by 6-OHDA in rats. Neuroscience 2016, 318, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.M.; Stroebel, B.M.; Faltesek, K.A.; Terai, K.; Haase, L.; Knutzen, K.E.; Kosyakovsky, J.; Bowe, T.J.; Fuller, A.K.; Frey, W.H.; et al. Intranasal delivery of low-dose insulin ameliorates motor dysfunction and dopaminergic cell death in a 6-OHDA rat model of Parkinson’s Disease. Neurosci. Lett. 2020, 714, 134567. [Google Scholar] [CrossRef] [PubMed]
- Iravanpour, F.; Dargahi, L.; Rezaei, M.; Haghani, M.; Heidari, R.; Valian, N.; Ahmadiani, A. Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson’s disease. CNS Neurosci. Ther. 2021, 27, 308–319. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, J.C.; Carr, K.D.; Rice, M.E. Actions and Consequences of Insulin in the Striatum. Biomolecules 2023, 13, 518. https://doi.org/10.3390/biom13030518
Patel JC, Carr KD, Rice ME. Actions and Consequences of Insulin in the Striatum. Biomolecules. 2023; 13(3):518. https://doi.org/10.3390/biom13030518
Chicago/Turabian StylePatel, Jyoti C., Kenneth D. Carr, and Margaret E. Rice. 2023. "Actions and Consequences of Insulin in the Striatum" Biomolecules 13, no. 3: 518. https://doi.org/10.3390/biom13030518
APA StylePatel, J. C., Carr, K. D., & Rice, M. E. (2023). Actions and Consequences of Insulin in the Striatum. Biomolecules, 13(3), 518. https://doi.org/10.3390/biom13030518