
Emerging Quantitative Biochemical, Structural, and Biophysical Methods for Studying Ribosome and Protein–RNA Complex Assembly

Abstract
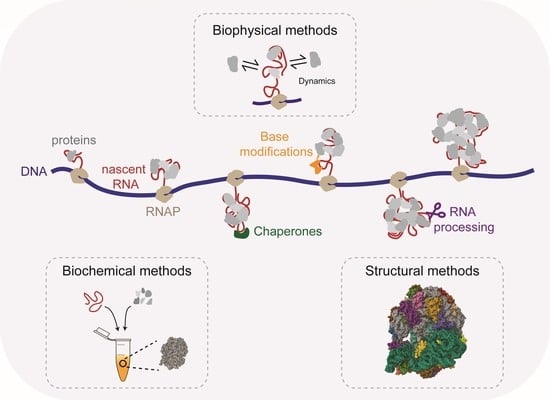
:
{kind=link}
{kind=link}
{kind=link}
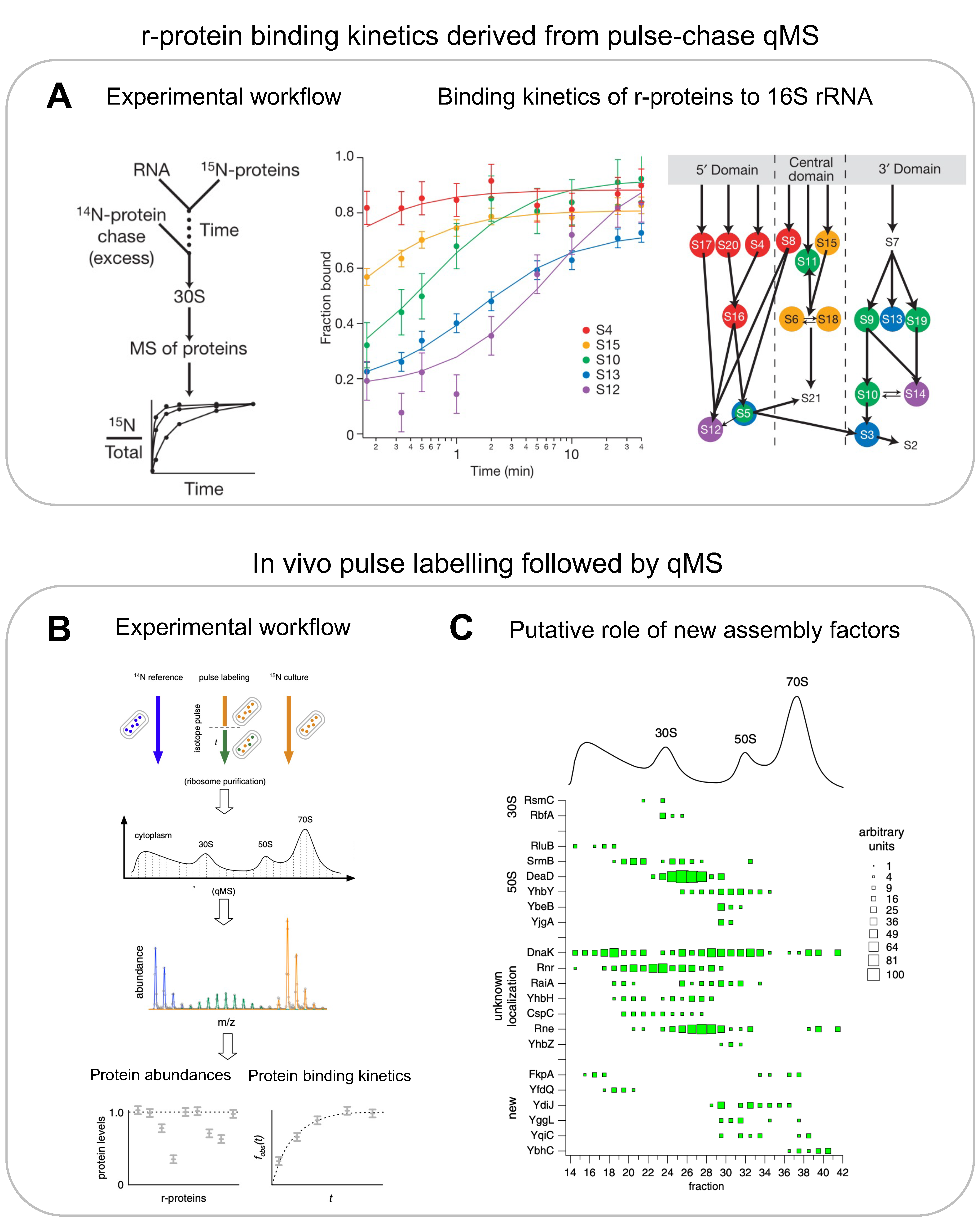{kind=link}
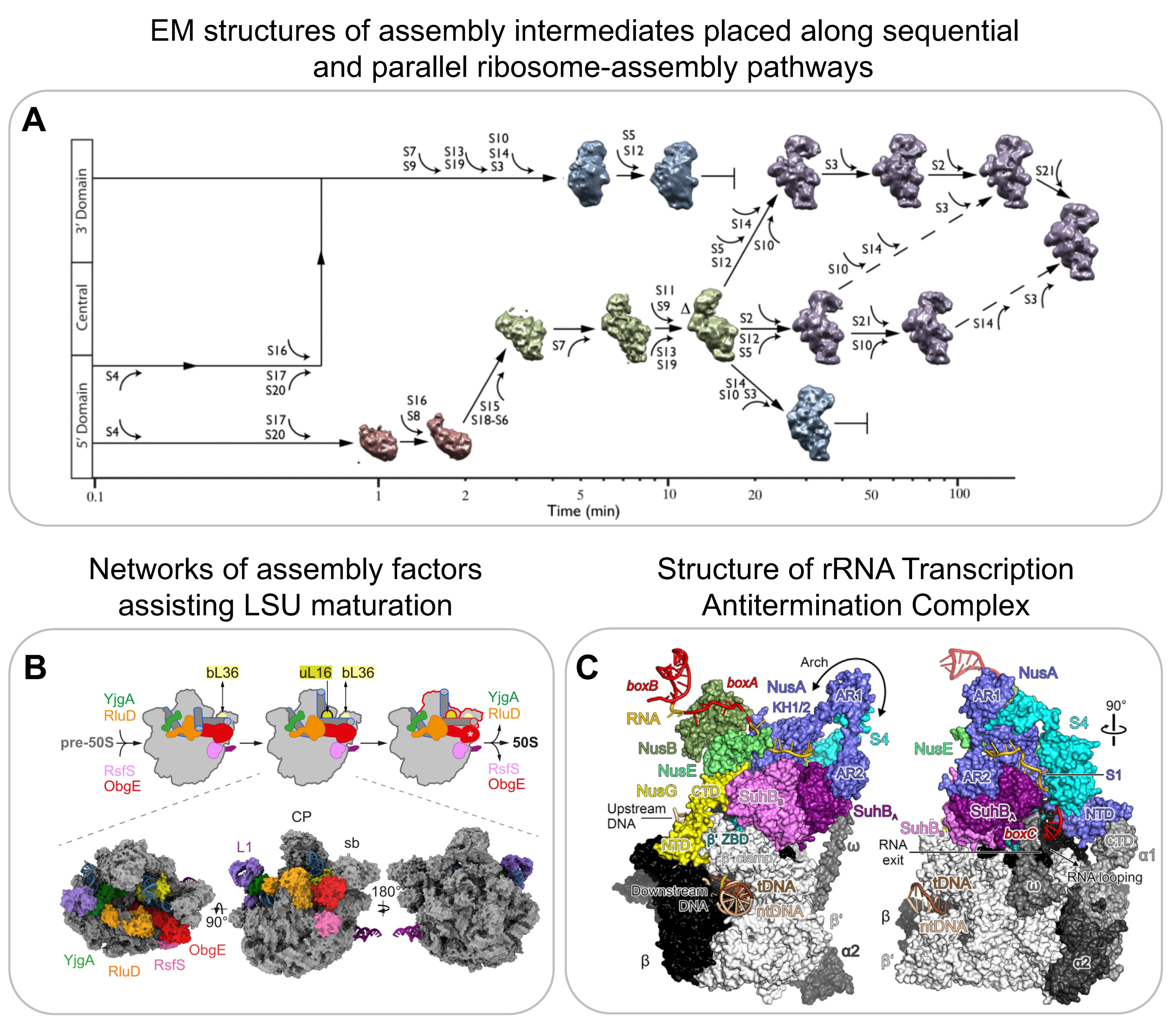{kind=link}
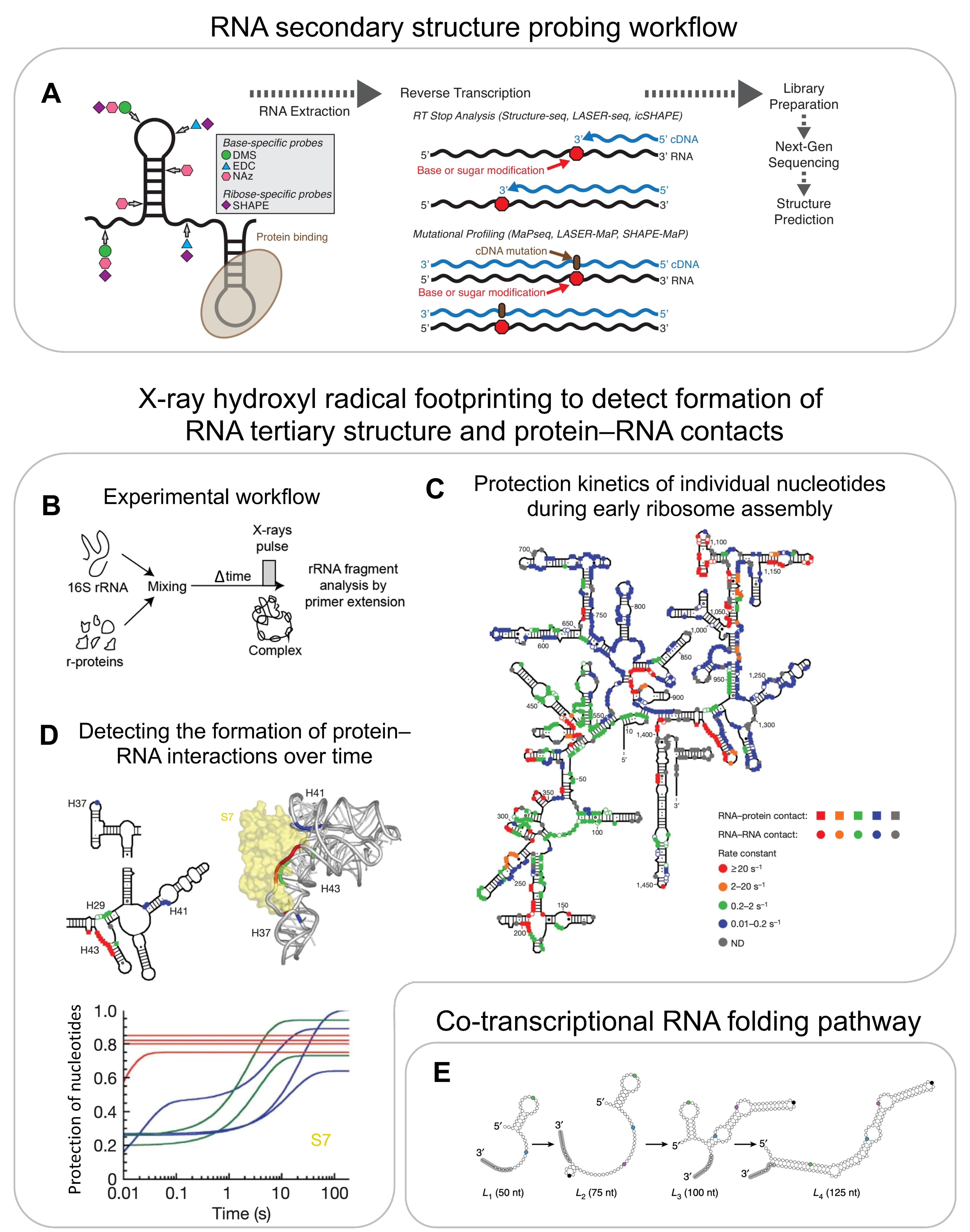{kind=link}
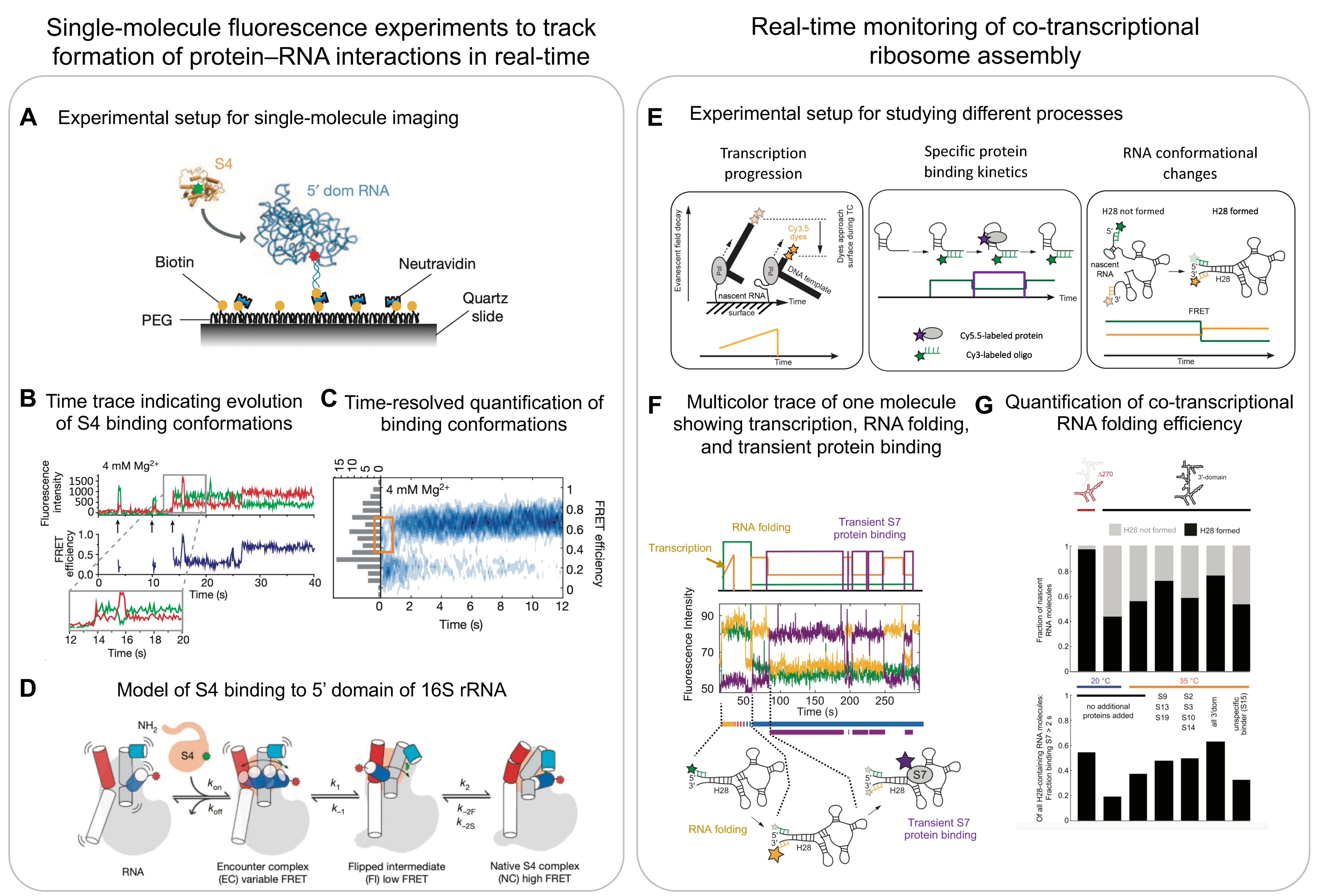{kind=link}
{kind=link}
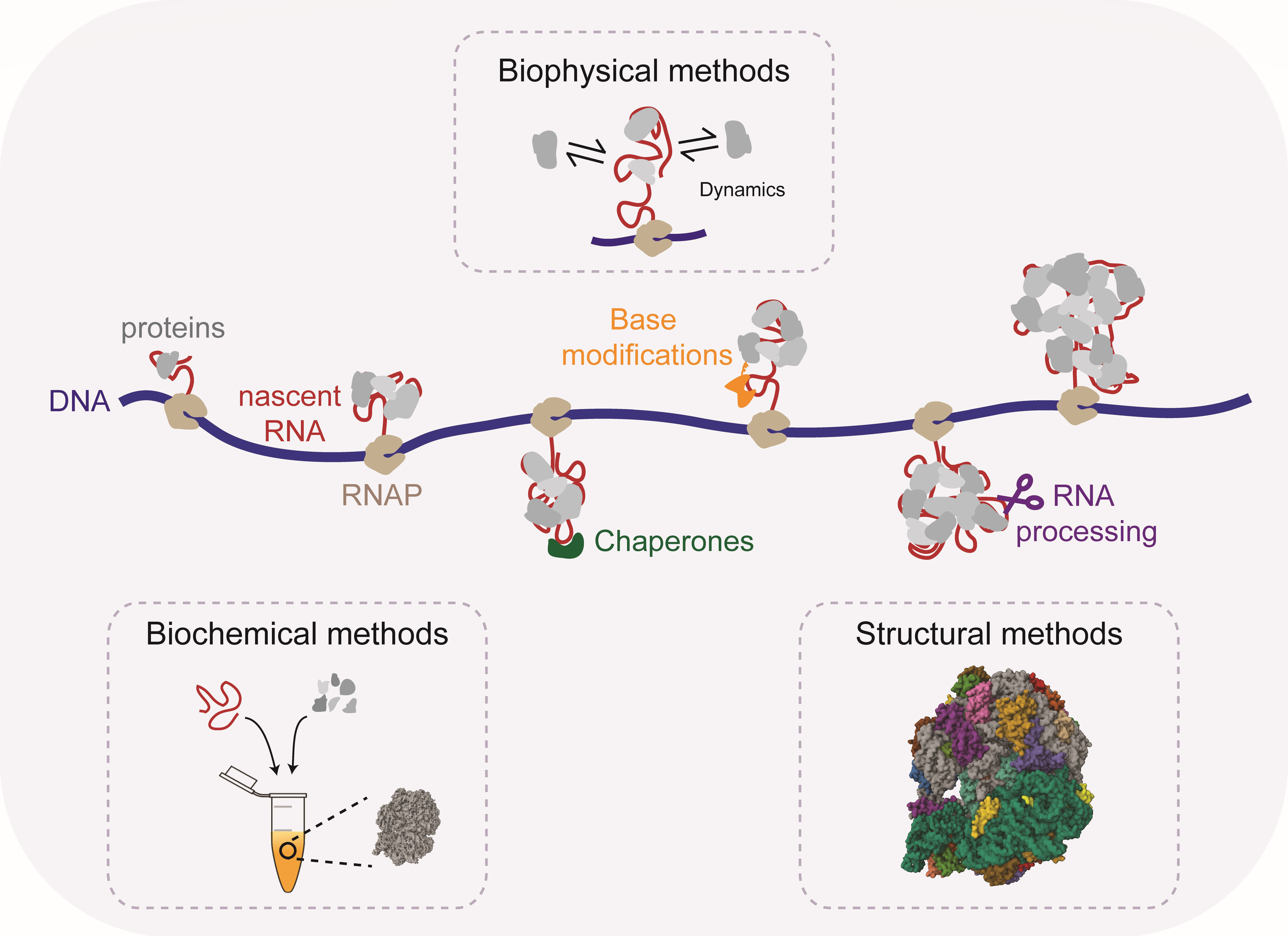
1. Introduction
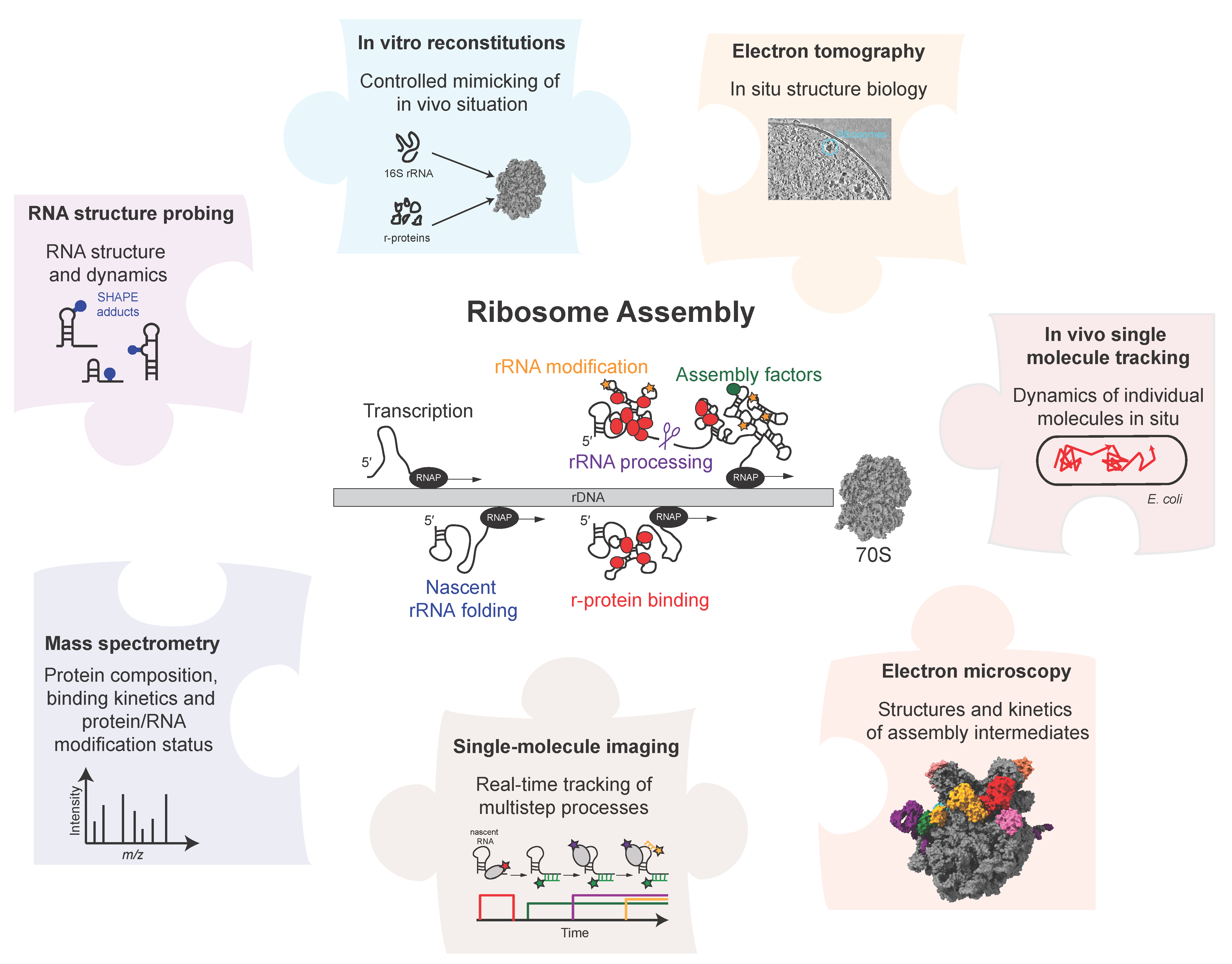
2. Biochemical Reconstitutions
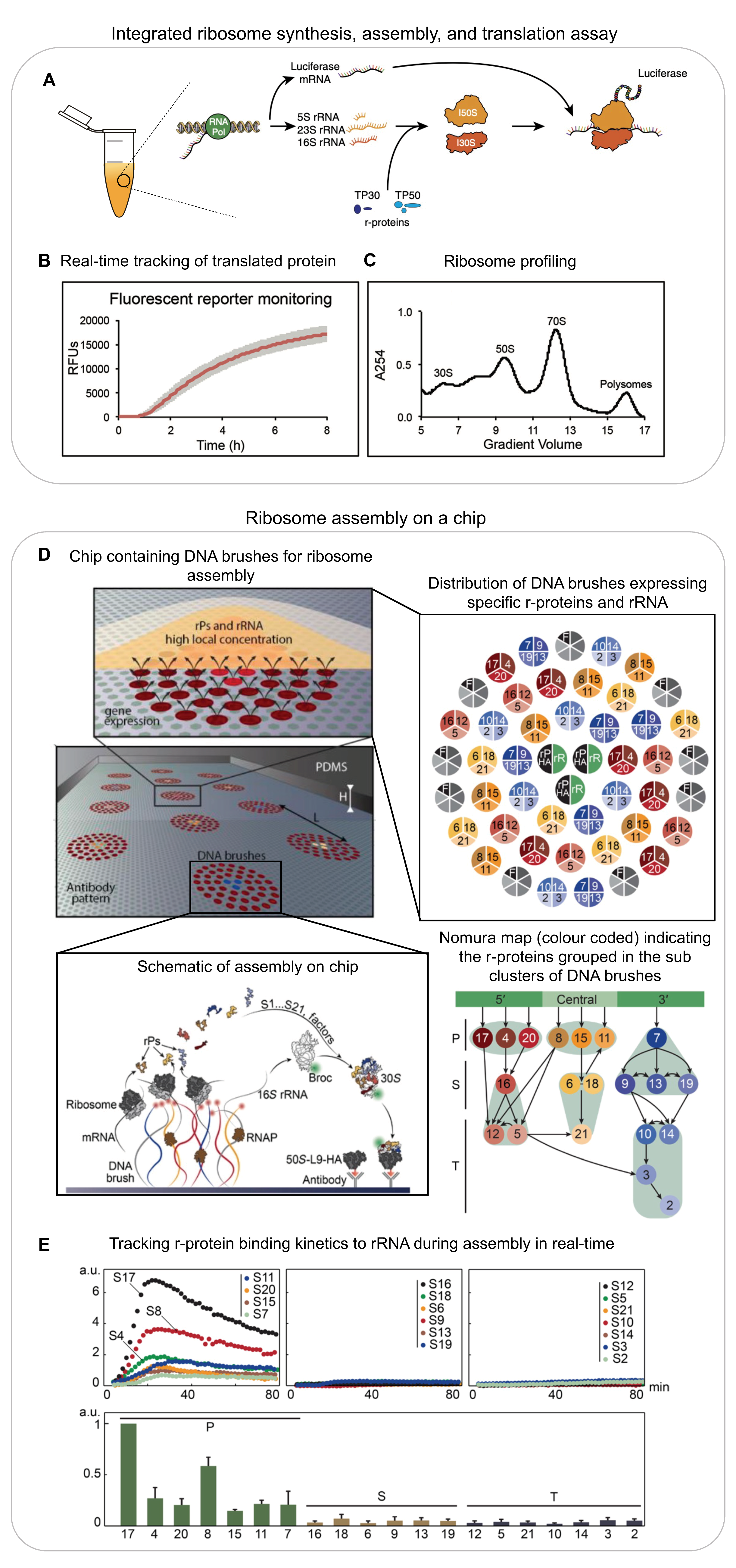
2.1. In Vitro Reconstitutions
2.2. In Vivo Mimicry
3. Mass Spectrometry
3.1. In Vitro Mass Spectrometry for r-Proteins
3.2. In Vivo Mass Spectrometry for r-Proteins and Assembly Factors
3.3. In Vivo Mass Spectrometry for RNA Modifications
4. Electron Microscopy
5. RNA Structure Probing
5.1. In Vitro RNA Structure Probing

5.2. Co-Transcriptional RNA Structure Probing
5.3. In Vivo RNA Structure Probing
6. Single-Molecule Methods
6.1. Multicolour Single-Molecule Fluorescence Microscopy
6.2. Co-Transcriptional Single-Molecule Imaging
6.3. Optical Tweezers
7. Integrative Methods
8. Future Methods
8.1. Multicolour and Multiscale Single-Molecule Methods

8.2. In Vivo Single-Molecule Tracking
8.3. Cryo–Electron Tomography
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shajani, Z.; Sykes, M.T.; Williamson, J.R. Assembly of bacterial ribosomes. Annu. Rev. Biochem. 2011, 80, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H. The assembly of prokaryotic ribosomes. Biochimie 1991, 73, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Said, N.; Loll, B.; Wahl, M.C. Structural basis for the function of SuhB as a transcription factor in ribosomal RNA synthesis. Nucleic Acids Res. 2019, 47, 6488–6503. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bubunenko, M.; Smith, C.; Abbott, D.M.; Stringer, A.M.; Shi, R.; Court, D.L.; Wade, J.T. SuhB Associates with Nus Factors To Facilitate 30S Ribosome Biogenesis in Escherichia coli. MBio 2016, 7, e00114. [Google Scholar] [CrossRef]
- Torres, M.; Condon, C.; Balada, J.M.; Squires, C.; Squires, C.L. Ribosomal protein S4 is a transcription factor with properties remarkably similar to NusA, a protein involved in both non-ribosomal and ribosomal RNA antitermination. EMBO J. 2001, 20, 3811–3820. [Google Scholar] [CrossRef]
- Li, Z.; Pandit, S.; Deutscher, M.P. Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA 1999, 5, 139–146. [Google Scholar] [CrossRef]
- Li, Z.; Pandit, S.; Deutscher, M.P. RNase G (CafA protein) and RNase E are both required for the 5’ maturation of 16S ribosomal RNA. EMBO J. 1999, 18, 2878–2885. [Google Scholar] [CrossRef]
- Ginsburg, D.; Steitz, J.A. The 30 S ribosomal precursor RNA from Escherichia coli. A primary transcript containing 23 S, 16 S, and 5 S sequences. J. Biol. Chem. 1975, 250, 5647–5654. [Google Scholar] [CrossRef]
- King, T.C.; Schlessinger, D. S1 nuclease mapping analysis of ribosomal RNA processing in wild type and processing deficient Escherichia coli. J. Biol. Chem. 1983, 258, 12034–12042. [Google Scholar] [CrossRef]
- Rodgers, M.L.; Woodson, S.A. Transcription Increases the Cooperativity of Ribonucleoprotein Assembly. Cell 2019, 179, 1370–1381.e1312. [Google Scholar] [CrossRef]
- Duss, O.; Stepanyuk, G.A.; Puglisi, J.D.; Williamson, J.R. Transient Protein-RNA Interactions Guide Nascent Ribosomal RNA Folding. Cell 2019, 179, 1357–1369.e1316. [Google Scholar] [CrossRef] [PubMed]
- Herschlag, D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995, 270, 20871–20874. [Google Scholar] [CrossRef] [PubMed]
- Adilakshmi, T.; Ramaswamy, P.; Woodson, S.A. Protein-independent folding pathway of the 16S rRNA 5’ domain. J. Mol. Biol. 2005, 351, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Powers, T.; Daubresse, G.; Noller, H.F. Dynamics of in vitro assembly of 16 S rRNA into 30 S ribosomal subunits. J. Mol. Biol. 1993, 232, 362–374. [Google Scholar] [CrossRef]
- Talkington, M.W.; Siuzdak, G.; Williamson, J.R. An assembly landscape for the 30S ribosomal subunit. Nature 2005, 438, 628–632. [Google Scholar] [CrossRef]
- Held, W.A.; Ballou, B.; Mizushima, S.; Nomura, M. Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J. Biol. Chem. 1974, 249, 3103–3111. [Google Scholar] [CrossRef]
- Mizushima, S.; Nomura, M. Assembly mapping of 30S ribosomal proteins from E. coli. Nature 1970, 226, 1214. [Google Scholar] [CrossRef]
- Rohl, R.; Nierhaus, K.H. Assembly map of the large subunit (50S) of Escherichia coli ribosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 729–733. [Google Scholar] [CrossRef]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Rodgers, M.L.; Woodson, S.A. A roadmap for rRNA folding and assembly during transcription. Trends Biochem. Sci. 2021, 46, 889–901. [Google Scholar] [CrossRef]
- Britton, R.A. Role of GTPases in bacterial ribosome assembly. Annu. Rev. Microbiol. 2009, 63, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Williamson, J.R. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013, 425, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, L. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J. Mol. Biol. 1975, 92, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Williamson, J.R. Structure and dynamics of bacterial ribosome biogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160181. [Google Scholar] [CrossRef]
- Naganathan, A.; Culver, G.M. Interdependency and Redundancy Add Complexity and Resilience to Biogenesis of Bacterial Ribosomes. Annu. Rev. Microbiol. 2022, 76, 193–210. [Google Scholar] [CrossRef]
- Oborska-Oplova, M.; Gerhardy, S.; Panse, V.G. Orchestrating ribosomal RNA folding during ribosome assembly. Bioessays 2022, 44, e2200066. [Google Scholar] [CrossRef]
- Sykes, M.T.; Williamson, J.R. A complex assembly landscape for the 30S ribosomal subunit. Annu. Rev. Biophys. 2009, 38, 197–215. [Google Scholar] [CrossRef]
- Williamson, J.R. Biophysical studies of bacterial ribosome assembly. Curr. Opin. Struct. Biol. 2008, 18, 299–304. [Google Scholar] [CrossRef]
- Woodson, S.A. RNA folding and ribosome assembly. Curr. Opin. Chem. Biol. 2008, 12, 667–673. [Google Scholar] [CrossRef]
- Woodson, S.A. RNA folding pathways and the self-assembly of ribosomes. Acc. Chem. Res. 2011, 44, 1312–1319. [Google Scholar] [CrossRef]
- Xue, L.; Lenz, S.; Zimmermann-Kogadeeva, M.; Tegunov, D.; Cramer, P.; Bork, P.; Rappsilber, J.; Mahamid, J. Visualizing translation dynamics at atomic detail inside a bacterial cell. Nature 2022, 610, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Culver, G.M.; Noller, H.F. Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA 1999, 5, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, R.; Masuda, K.; Shimojo, M.; Kanamori, T.; Ueda, T.; Shimizu, Y. In vitro reconstitution of the Escherichia coli 70S ribosome with a full set of recombinant ribosomal proteins. J. Biochem. 2022, 171, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H.; Dohme, F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 4713–4717. [Google Scholar] [CrossRef]
- Rheinberger, H.J.; Nierhaus, K.H. Partial release of AcPhe-Phe-tRNA from ribosomes during poly(U)-dependent poly(Phe) synthesis and the effects of chloramphenicol. Eur. J. Biochem. 1990, 193, 643–650. [Google Scholar] [CrossRef]
- Tamaru, D.; Amikura, K.; Shimizu, Y.; Nierhaus, K.H.; Ueda, T. Reconstitution of 30S ribosomal subunits in vitro using ribosome biogenesis factors. RNA 2018, 24, 1512–1519. [Google Scholar] [CrossRef]
- Semrad, K.; Green, R. Osmolytes stimulate the reconstitution of functional 50S ribosomes from in vitro transcripts of Escherichia coli 23S rRNA. RNA 2002, 8, 401–411. [Google Scholar] [CrossRef]
- Maki, J.A.; Culver, G.M. Recent developments in factor-facilitated ribosome assembly. Methods 2005, 36, 313–320. [Google Scholar] [CrossRef]
- Nomura, M.; Traub, P.; Guthrie, C.; Nashimoto, H. The assembly of ribosomes. J. Cell Physiol. 1969, 74 (Suppl. S1), 241. [Google Scholar] [CrossRef]
- Jewett, M.C.; Fritz, B.R.; Timmerman, L.E.; Church, G.M. In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol. Syst. Biol. 2013, 9, 678. [Google Scholar] [CrossRef]
- Miller, O.L., Jr.; Hamkalo, B.A.; Thomas, C.A., Jr. Visualization of bacterial genes in action. Science 1970, 169, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.L., Jr.; Beatty, B.R. Visualization of nucleolar genes. Science 1969, 164, 955–957. [Google Scholar] [CrossRef]
- Hofmann, S.; Miller, O.L., Jr. Visualization of ribosomal ribonucleic acid synthesis in a ribonuclease III-Deficient strain of Escherichia coli. J. Bacteriol. 1977, 132, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Gotta, S.L.; Miller, O.L., Jr.; French, S.L. rRNA transcription rate in Escherichia coli. J. Bacteriol. 1991, 173, 6647–6649. [Google Scholar] [CrossRef] [PubMed]
- Fritz, B.R.; Jewett, M.C. The impact of transcriptional tuning on in vitro integrated rRNA transcription and ribosome construction. Nucleic Acids Res. 2014, 42, 6774–6785. [Google Scholar] [CrossRef]
- Dong, X.; Doerfel, L.K.; Sheng, K.; Rabuck-Gibbons, J.N.; Popova, A.M.; Lyumkis, D.; Williamson, J.R. Near-physiological in vitro assembly of 50S ribosomes involves parallel pathways. Nucleic Acids Res. 2023, 51, 2862–2876. [Google Scholar] [CrossRef]
- Fritz, B.R.; Jamil, O.K.; Jewett, M.C. Implications of macromolecular crowding and reducing conditions for in vitro ribosome construction. Nucleic Acids Res. 2015, 43, 4774–4784. [Google Scholar] [CrossRef]
- Shimojo, M.; Amikura, K.; Masuda, K.; Kanamori, T.; Ueda, T.; Shimizu, Y. In vitro reconstitution of functional small ribosomal subunit assembly for comprehensive analysis of ribosomal elements in E. coli. Commun. Biol. 2020, 3, 142. [Google Scholar] [CrossRef]
- Levy, M.; Falkovich, R.; Daube, S.S.; Bar-Ziv, R.H. Autonomous synthesis and assembly of a ribosomal subunit on a chip. Sci. Adv. 2020, 6, eaaz6020. [Google Scholar] [CrossRef]
- d’Aquino, A.E.; Azim, T.; Aleksashin, N.A.; Hockenberry, A.J.; Kruger, A.; Jewett, M.C. Mutational characterization and mapping of the 70S ribosome active site. Nucleic Acids Res. 2020, 48, 2777–2789. [Google Scholar] [CrossRef]
- Liu, Y.; Davis, R.G.; Thomas, P.M.; Kelleher, N.L.; Jewett, M.C. In vitro-Constructed Ribosomes Enable Multi-site Incorporation of Noncanonical Amino Acids into Proteins. Biochemistry 2021, 60, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, M.J.; Fritz, B.R.; Yoesep, D.J.; Kim, D.S.; Carlson, E.D.; Jewett, M.C. In vitro ribosome synthesis and evolution through ribosome display. Nat. Commun. 2020, 11, 1108. [Google Scholar] [CrossRef] [PubMed]
- Bunner, A.E.; Beck, A.H.; Williamson, J.R. Kinetic cooperativity in Escherichia coli 30S ribosomal subunit reconstitution reveals additional complexity in the assembly landscape. Proc. Natl. Acad. Sci. USA 2010, 107, 5417–5422. [Google Scholar] [CrossRef] [PubMed]
- Bunner, A.E.; Nord, S.; Wikstrom, P.M.; Williamson, J.R. The effect of ribosome assembly cofactors on in vitro 30S subunit reconstitution. J. Mol. Biol. 2010, 398, 1–7. [Google Scholar] [CrossRef]
- Nord, S.; Bylund, G.O.; Lovgren, J.M.; Wikstrom, P.M. The RimP protein is important for maturation of the 30S ribosomal subunit. J. Mol. Biol. 2009, 386, 742–753. [Google Scholar] [CrossRef]
- Bylund, G.O.; Persson, B.C.; Lundberg, L.A.; Wikstrom, P.M. A novel ribosome-associated protein is important for efficient translation in Escherichia coli. J. Bacteriol. 1997, 179, 4567–4574. [Google Scholar] [CrossRef]
- Inoue, K.; Alsina, J.; Chen, J.; Inouye, M. Suppression of defective ribosome assembly in a rbfA deletion mutant by overexpression of Era, an essential GTPase in Escherichia coli. Mol. Microbiol. 2003, 48, 1005–1016. [Google Scholar] [CrossRef]
- Gibbs, M.R.; Moon, K.M.; Chen, M.; Balakrishnan, R.; Foster, L.J.; Fredrick, K. Conserved GTPase LepA (Elongation Factor 4) functions in biogenesis of the 30S subunit of the 70S ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, 980–985. [Google Scholar] [CrossRef]
- Clatterbuck Soper, S.F.; Dator, R.P.; Limbach, P.A.; Woodson, S.A. In vivo X-ray footprinting of pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol. Cell 2013, 52, 506–516. [Google Scholar] [CrossRef]
- Sailer, C.; Jansen, J.; Sekulski, K.; Cruz, V.E.; Erzberger, J.P.; Stengel, F. A comprehensive landscape of 60S ribosome biogenesis factors. Cell Rep. 2022, 38, 110353. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 2007, 425, 21–53. [Google Scholar] [CrossRef] [PubMed]
- Siibak, T.; Remme, J. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA 2010, 16, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Kellner, S.; Ochel, A.; Thuring, K.; Spenkuch, F.; Neumann, J.; Sharma, S.; Entian, K.D.; Schneider, D.; Helm, M. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 2014, 42, e142. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.M.; Williamson, J.R. Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J. Am. Chem. Soc. 2014, 136, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Arai, T.; Suzuki, T. Depletion of S-adenosylmethionine impacts on ribosome biogenesis through hypomodification of a single rRNA methylation. Nucleic Acids Res. 2019, 47, 4226–4239. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Mulder, A.M.; Yoshioka, C.; Beck, A.H.; Bunner, A.E.; Milligan, R.A.; Potter, C.S.; Carragher, B.; Williamson, J.R. Visualizing ribosome biogenesis: Parallel assembly pathways for the 30S subunit. Science 2010, 330, 673–677. [Google Scholar] [CrossRef]
- Nikolay, R.; Hilal, T.; Schmidt, S.; Qin, B.; Schwefel, D.; Vieira-Vieira, C.H.; Mielke, T.; Burger, J.; Loerke, J.; Amikura, K.; et al. Snapshots of native pre-50S ribosomes reveal a biogenesis factor network and evolutionary specialization. Mol. Cell 2021, 81, 1200–1215.e1209. [Google Scholar] [CrossRef]
- Huang, Y.H.; Hilal, T.; Loll, B.; Burger, J.; Mielke, T.; Bottcher, C.; Said, N.; Wahl, M.C. Structure-Based Mechanisms of a Molecular RNA Polymerase/Chaperone Machine Required for Ribosome Biosynthesis. Mol. Cell 2020, 79, 1024–1036.e1025. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Callaway, E. ‘It opens up a whole new universe’: Revolutionary microscopy technique sees individual atoms for first time. Nature 2020, 582, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Nikolay, R.; Hilal, T.; Qin, B.; Mielke, T.; Burger, J.; Loerke, J.; Textoris-Taube, K.; Nierhaus, K.H.; Spahn, C.M.T. Structural Visualization of the Formation and Activation of the 50S Ribosomal Subunit during In Vitro Reconstitution. Mol. Cell 2018, 70, 881–893.e883. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Tan, Y.Z.; Carragher, B.; Potter, C.S.; Lyumkis, D.; Williamson, J.R. Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell 2016, 167, 1610–1622.e1615. [Google Scholar] [CrossRef] [PubMed]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, W.; Ge, X.; Yan, K.; Mandava, C.S.; Sanyal, S.; Gao, N. Loss of a single methylation in 23S rRNA delays 50S assembly at multiple late stages and impairs translation initiation and elongation. Proc. Natl. Acad. Sci. USA 2020, 117, 15609–15619. [Google Scholar] [CrossRef] [PubMed]
- Rabuck-Gibbons, J.N.; Popova, A.M.; Greene, E.M.; Cervantes, C.F.; Lyumkis, D.; Williamson, J.R. SrmB Rescues Trapped Ribosome Assembly Intermediates. J. Mol. Biol. 2020, 432, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Razi, A.; Davis, J.H.; Hao, Y.; Jahagirdar, D.; Thurlow, B.; Basu, K.; Jain, N.; Gomez-Blanco, J.; Britton, R.A.; Vargas, J.; et al. Role of Era in assembly and homeostasis of the ribosomal small subunit. Nucleic Acids Res. 2019, 47, 8301–8317. [Google Scholar] [CrossRef]
- Thurlow, B.; Davis, J.H.; Leong, V.; Moraes, T.F.; Williamson, J.R.; Ortega, J. Binding properties of YjeQ (RsgA), RbfA, RimM and Era to assembly intermediates of the 30S subunit. Nucleic Acids Res. 2016, 44, 9918–9932. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Goto, S.; Chen, Y.; Feng, B.; Xu, Y.; Muto, A.; Himeno, H.; Deng, H.; Lei, J.; Gao, N. Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res. 2013, 41, 2609–2620. [Google Scholar] [CrossRef]
- Jomaa, A.; Stewart, G.; Martin-Benito, J.; Zielke, R.; Campbell, T.L.; Maddock, J.R.; Brown, E.D.; Ortega, J. Understanding ribosome assembly: The structure of in vivo assembled immature 30S subunits revealed by cryo-electron microscopy. RNA 2011, 17, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Leong, V.; Kent, M.; Jomaa, A.; Ortega, J. Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA 2013, 19, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Boehringer, D.; O’Farrell, H.C.; Rife, J.P.; Ban, N. Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J. Biol. Chem. 2012, 287, 10453–10459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, K.; Zhang, Y.; Li, N.; Ma, C.; Li, Z.; Zhang, Y.; Feng, B.; Liu, J.; Sun, Y.; et al. Structural insights into the function of a unique tandem GTPase EngA in bacterial ribosome assembly. Nucleic Acids Res. 2014, 42, 13430–13439. [Google Scholar] [CrossRef] [PubMed]
- Bubunenko, M.; Court, D.L.; Al Refaii, A.; Saxena, S.; Korepanov, A.; Friedman, D.I.; Gottesman, M.E.; Alix, J.H. Nus transcription elongation factors and RNase III modulate small ribosome subunit biogenesis in Escherichia coli. Mol. Microbiol. 2013, 87, 382–393. [Google Scholar] [CrossRef]
- Redko, Y.; Condon, C. Ribosomal protein L3 bound to 23S precursor rRNA stimulates its maturation by Mini-III ribonuclease. Mol. Microbiol. 2009, 71, 1145–1154. [Google Scholar] [CrossRef]
- Stahl, D.A.; Pace, B.; Marsh, T.; Pace, N.R. The ribonucleoprotein substrate for a ribosomal RNA-processing nuclease. J. Biol. Chem. 1984, 259, 11448–11453. [Google Scholar] [CrossRef]
- Oerum, S.; Dendooven, T.; Catala, M.; Gilet, L.; Degut, C.; Trinquier, A.; Bourguet, M.; Barraud, P.; Cianferani, S.; Luisi, B.F.; et al. Structures of B. subtilis Maturation RNases Captured on 50S Ribosome with Pre-rRNAs. Mol. Cell 2020, 80, 227–236.e225. [Google Scholar] [CrossRef]
- Stiegler, P.; Carbon, P.; Zuker, M.; Ebel, J.P.; Ehresmann, C. Structural organization of the 16S ribosomal RNA from E. coli. Topography and secondary structure. Nucleic Acids Res. 1981, 9, 2153–2172. [Google Scholar] [CrossRef]
- Spitale, R.C.; Incarnato, D. Probing the dynamic RNA structurome and its functions. Nat. Rev. Genet. 2023, 24, 178–196. [Google Scholar] [CrossRef]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2009, 106, 97–102. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Adilakshmi, T.; Bellur, D.L.; Woodson, S.A. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 2008, 455, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Powers, T.; Changchien, L.M.; Noller, H.F. RNA-protein interactions in 30S ribosomal subunits: Folding and function of 16S rRNA. Science 1989, 244, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Abeysirigunawarden, S.C.; Chen, K.; Mayerle, M.; Ragunathan, K.; Luthey-Schulten, Z.; Ha, T.; Woodson, S.A. Protein-guided RNA dynamics during early ribosome assembly. Nature 2014, 506, 334–338. [Google Scholar] [CrossRef]
- Duss, O.; Stepanyuk, G.A.; Grot, A.; O’Leary, S.E.; Puglisi, J.D.; Williamson, J.R. Real-time assembly of ribonucleoprotein complexes on nascent RNA transcripts. Nat. Commun. 2018, 9, 5087. [Google Scholar] [CrossRef]
- Olson, S.W.; Turner, A.W.; Arney, J.W.; Saleem, I.; Weidmann, C.A.; Margolis, D.M.; Weeks, K.M.; Mustoe, A.M. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Mol. Cell 2022, 82, 1708–1723.e1710. [Google Scholar] [CrossRef]
- Morandi, E.; Manfredonia, I.; Simon, L.M.; Anselmi, F.; van Hemert, M.J.; Oliviero, S.; Incarnato, D. Genome-scale deconvolution of RNA structure ensembles. Nat. Methods 2021, 18, 249–252. [Google Scholar] [CrossRef]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; McIntosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef]
- Homan, P.J.; Favorov, O.V.; Lavender, C.A.; Kursun, O.; Ge, X.; Busan, S.; Dokholyan, N.V.; Weeks, K.M. Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA 2014, 111, 13858–13863. [Google Scholar] [CrossRef]
- Khoroshkin, M.; Asarnow, D.; Navickas, A.; Winters, A.; Yu, J.; Zhou, S.K.; Zhou, S.; Palka, C.; Fish, L.; Ansel, K.M.; et al. A systematic search for RNA structural switches across the human transcriptome. bioRxiv 2023. [Google Scholar] [CrossRef]
- Mitchell, D., 3rd; Assmann, S.M.; Bevilacqua, P.C. Probing RNA structure in vivo. Curr. Opin. Struct. Biol. 2019, 59, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Watters, K.E.; Strobel, E.J.; Yu, A.M.; Lis, J.T.; Lucks, J.B. Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol. 2016, 23, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Lewicki, B.T.; Margus, T.; Remme, J.; Nierhaus, K.H. Coupling of rRNA transcription and ribosomal assembly in vivo. Formation of active ribosomal subunits in Escherichia coli requires transcription of rRNA genes by host RNA polymerase which cannot be replaced by bacteriophage T7 RNA polymerase. J. Mol. Biol. 1993, 231, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Hulscher, R.M.; Bohon, J.; Rappe, M.C.; Gupta, S.; D’Mello, R.; Sullivan, M.; Ralston, C.Y.; Chance, M.R.; Woodson, S.A. Probing the structure of ribosome assembly intermediates in vivo using DMS and hydroxyl radical footprinting. Methods 2016, 103, 49–56. [Google Scholar] [CrossRef]
- Mortimer, S.A.; Weeks, K.M. Time-resolved RNA SHAPE chemistry. J. Am. Chem. Soc. 2008, 130, 16178–16180. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Feng, X.A.; Poyton, M.F.; Ha, T. Multicolor single-molecule FRET for DNA and RNA processes. Curr. Opin. Struct. Biol. 2021, 70, 26–33. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359, eaan1133. [Google Scholar] [CrossRef]
- Ha, T.; Zhuang, X.; Kim, H.D.; Orr, J.W.; Williamson, J.R.; Chu, S. Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 9077–9082. [Google Scholar] [CrossRef]
- Abeysirigunawardena, S.C.; Kim, H.; Lai, J.; Ragunathan, K.; Rappe, M.C.; Luthey-Schulten, Z.; Ha, T.; Woodson, S.A. Evolution of protein-coupled RNA dynamics during hierarchical assembly of ribosomal complexes. Nat. Commun. 2017, 8, 492. [Google Scholar] [CrossRef]
- Lai, D.; Proctor, J.R.; Meyer, I.M. On the importance of cotranscriptional RNA structure formation. RNA 2013, 19, 1461–1473. [Google Scholar] [CrossRef]
- Zhang, J.; Landick, R. A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends. Biochem. Sci. 2016, 41, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Uhm, H.; Kang, W.; Ha, K.S.; Kang, C.; Hohng, S. Single-molecule FRET studies on the cotranscriptional folding of a thiamine pyrophosphate riboswitch. Proc. Natl. Acad. Sci. USA 2018, 115, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Chauvier, A.; St-Pierre, P.; Nadon, J.F.; Hien, E.D.M.; Perez-Gonzalez, C.; Eschbach, S.H.; Lamontagne, A.M.; Penedo, J.C.; Lafontaine, D.A. Monitoring RNA dynamics in native transcriptional complexes. Proc. Natl. Acad. Sci. USA 2021, 118, e2106564118. [Google Scholar] [CrossRef] [PubMed]
- Hua, B.; Panja, S.; Wang, Y.; Woodson, S.A.; Ha, T. Mimicking Co-Transcriptional RNA Folding Using a Superhelicase. J. Am. Chem. Soc. 2018, 140, 10067–10070. [Google Scholar] [CrossRef]
- Wang, M.D.; Schnitzer, M.J.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Force and velocity measured for single molecules of RNA polymerase. Science 1998, 282, 902–907. [Google Scholar] [CrossRef]
- Abbondanzieri, E.A.; Greenleaf, W.J.; Shaevitz, J.W.; Landick, R.; Block, S.M. Direct observation of base-pair stepping by RNA polymerase. Nature 2005, 438, 460–465. [Google Scholar] [CrossRef]
- Frieda, K.L.; Block, S.M. Direct observation of cotranscriptional folding in an adenine riboswitch. Science 2012, 338, 397–400. [Google Scholar] [CrossRef]
- Mangeol, P.; Bizebard, T.; Chiaruttini, C.; Dreyfus, M.; Springer, M.; Bockelmann, U. Probing ribosomal protein-RNA interactions with an external force. Proc. Natl. Acad. Sci. USA 2011, 108, 18272–18276. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schlessinger, D. Coregulation of processing and translation: Mature 5’ termini of Escherichia coli 23S ribosomal RNA form in polysomes. Proc. Natl. Acad. Sci. USA 1988, 85, 7144–7148. [Google Scholar] [CrossRef]
- Mangiarotti, G.; Turco, E.; Ponzetto, A.; Altruda, F. Precursor 16S RNA in active 30S ribosomes. Nature 1974, 247, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Lauer, S.M.; Balke, A.; Vieira-Vieira, C.H.; Burger, J.; Mielke, T.; Selbach, M.; Scheerer, P.; Spahn, C.M.T.; Nikolay, R. Cryo-EM captures early ribosome assembly in action. Nat. Commun. 2023, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Desai, V.P.; Frank, F.; Lee, A.; Righini, M.; Lancaster, L.; Noller, H.F.; Tinoco, I., Jr.; Bustamante, C. Co-temporal Force and Fluorescence Measurements Reveal a Ribosomal Gear Shift Mechanism of Translation Regulation by Structured mRNAs. Mol. Cell 2019, 75, 1007–1019.e1005. [Google Scholar] [CrossRef] [PubMed]
- C-TRAP® Optical Tweezers Fluorescence & Label-Free Microscopy. Available online: https://lumicks.com/products/c-trap-optical-tweezers-fluorescence-label-free-microscopy/ (accessed on 6 April 2023).
- Young, G.; Hundt, N.; Cole, D.; Fineberg, A.; Andrecka, J.; Tyler, A.; Olerinyova, A.; Ansari, A.; Marklund, E.G.; Collier, M.P.; et al. Quantitative mass imaging of single biological macromolecules. Science 2018, 360, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef]
- Begik, O.; Mattick, J.S.; Novoa, E.M. Exploring the epitranscriptome by native RNA sequencing. RNA 2022, 28, 1430–1439. [Google Scholar] [CrossRef]
- Begik, O.; Lucas, M.C.; Pryszcz, L.P.; Ramirez, J.M.; Medina, R.; Milenkovic, I.; Cruciani, S.; Liu, H.; Vieira, H.G.S.; Sas-Chen, A.; et al. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat. Biotechnol. 2021, 39, 1278–1291. [Google Scholar] [CrossRef]
- Hendra, C.; Pratanwanich, P.N.; Wan, Y.K.; Goh, W.S.S.; Thiery, A.; Goke, J. Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat. Methods 2022, 19, 1590–1598. [Google Scholar] [CrossRef]
- Mateos, P.A.; Sethi, A.J.; Ravindran, A.; Guarnacci, M.; Srivastava, A.; Xu, J.; Woodward, K.; Yuen, Z.W.S.; Mahmud, S.; Kanchi, M.; et al. Simultaneous identification of m6A and m5C reveals coordinated RNA modification at single-molecule resolution. bioRxiv 2023. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Lim, S.W.; Wang, J.X.; Lambert, F.R.P.; Tan, W.T.; Shen, Y.; Zhang, Y.; Kaewsapsak, P.; Li, C.; Ng, S.B.; et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 2021, 39, 336–346. [Google Scholar] [CrossRef]
- Wrzesinski, J.; Bakin, A.; Nurse, K.; Lane, B.G.; Ofengand, J. Purification, cloning, and properties of the 16S RNA pseudouridine 516 synthase from Escherichia coli. Biochemistry 1995, 34, 8904–8913. [Google Scholar] [CrossRef]
- O’Reilly, F.J.; Xue, L.; Graziadei, A.; Sinn, L.; Lenz, S.; Tegunov, D.; Blotz, C.; Singh, N.; Hagen, W.J.H.; Cramer, P.; et al. In-cell architecture of an actively transcribing-translating expressome. Science 2020, 369, 554–557. [Google Scholar] [CrossRef]
- Ladouceur, A.M.; Parmar, B.S.; Biedzinski, S.; Wall, J.; Tope, S.G.; Cohn, D.; Kim, A.; Soubry, N.; Reyes-Lamothe, R.; Weber, S.C. Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc. Natl. Acad. Sci. USA 2020, 117, 18540–18549. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffie, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Primers 2021, 1, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Moffitt, J.R.; Dempsey, G.T.; Xie, X.S.; Zhuang, X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc. Natl. Acad. Sci USA 2014, 111, 8452–8457. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Bohrer, C.H.; Bettridge, K.; Lagda, A.C.; Cagliero, C.; Jin, D.J.; Xiao, J. Spatial organization of RNA polymerase and its relationship with transcription in Escherichia coli. Proc. Natl. Acad. Sci. USA 2019, 116, 20115–20123. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J. Birth of Nucleolar Compartments: Phase Separation-Driven Ribosomal RNA Sorting and Processing. Mol. Cell 2019, 76, 694–696. [Google Scholar] [CrossRef]
- Ruland, J.A.; Kruger, A.M.; Dorner, K.; Bhatia, R.; Wirths, S.; Poetes, D.; Kutay, U.; Siebrasse, J.P.; Kubitscheck, U. Nuclear export of the pre-60S ribosomal subunit through single nuclear pores observed in real time. Nat. Commun. 2021, 12, 6211. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynna, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Iwanski, M.K.; Schentarra, E.M.; Heidebrecht, C.; Schmidt, L.; Heck, J.; Weihs, T.; Schnorrenberg, S.; Hoess, P.; Liu, S.; et al. Direct observation of motor protein stepping in living cells using MINFLUX. Science 2023, 379, 1010–1015. [Google Scholar] [CrossRef]
- Wolff, J.O.; Scheiderer, L.; Engelhardt, T.; Engelhardt, J.; Matthias, J.; Hell, S.W. MINFLUX dissects the unimpeded walking of kinesin-1. Science 2023, 379, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Turk, M.; Baumeister, W. The promise and the challenges of cryo-electron tomography. FEBS Lett. 2020, 594, 3243–3261. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, P.S.; Hou, Z.; Klumpe, S.; Khavnekar, S.; Beck, F.; Wilfling, F.; Plitzko, J.M.; Baumeister, W. In situ cryo-electron tomography reveals gradient organization of ribosome biogenesis in intact nucleoli. Nat. Commun. 2021, 12, 5364. [Google Scholar] [CrossRef]
- Earnest, T.M.; Lai, J.; Chen, K.; Hallock, M.J.; Williamson, J.R.; Luthey-Schulten, Z. Toward a Whole-Cell Model of Ribosome Biogenesis: Kinetic Modeling of SSU Assembly. Biophys. J. 2015, 109, 1117–1135. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gor, K.; Duss, O. Emerging Quantitative Biochemical, Structural, and Biophysical Methods for Studying Ribosome and Protein–RNA Complex Assembly. Biomolecules 2023, 13, 866. https://doi.org/10.3390/biom13050866
Gor K, Duss O. Emerging Quantitative Biochemical, Structural, and Biophysical Methods for Studying Ribosome and Protein–RNA Complex Assembly. Biomolecules. 2023; 13(5):866. https://doi.org/10.3390/biom13050866
Chicago/Turabian StyleGor, Kavan, and Olivier Duss. 2023. "Emerging Quantitative Biochemical, Structural, and Biophysical Methods for Studying Ribosome and Protein–RNA Complex Assembly" Biomolecules 13, no. 5: 866. https://doi.org/10.3390/biom13050866
APA StyleGor, K., & Duss, O. (2023). Emerging Quantitative Biochemical, Structural, and Biophysical Methods for Studying Ribosome and Protein–RNA Complex Assembly. Biomolecules, 13(5), 866. https://doi.org/10.3390/biom13050866