
RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, RNAi and the Generation of Stable Cell Lines
2.2. Western Blotting
2.3. Immunofluorescence
2.4. Gradient Analysis
2.5. RNA Analysis
3. Results
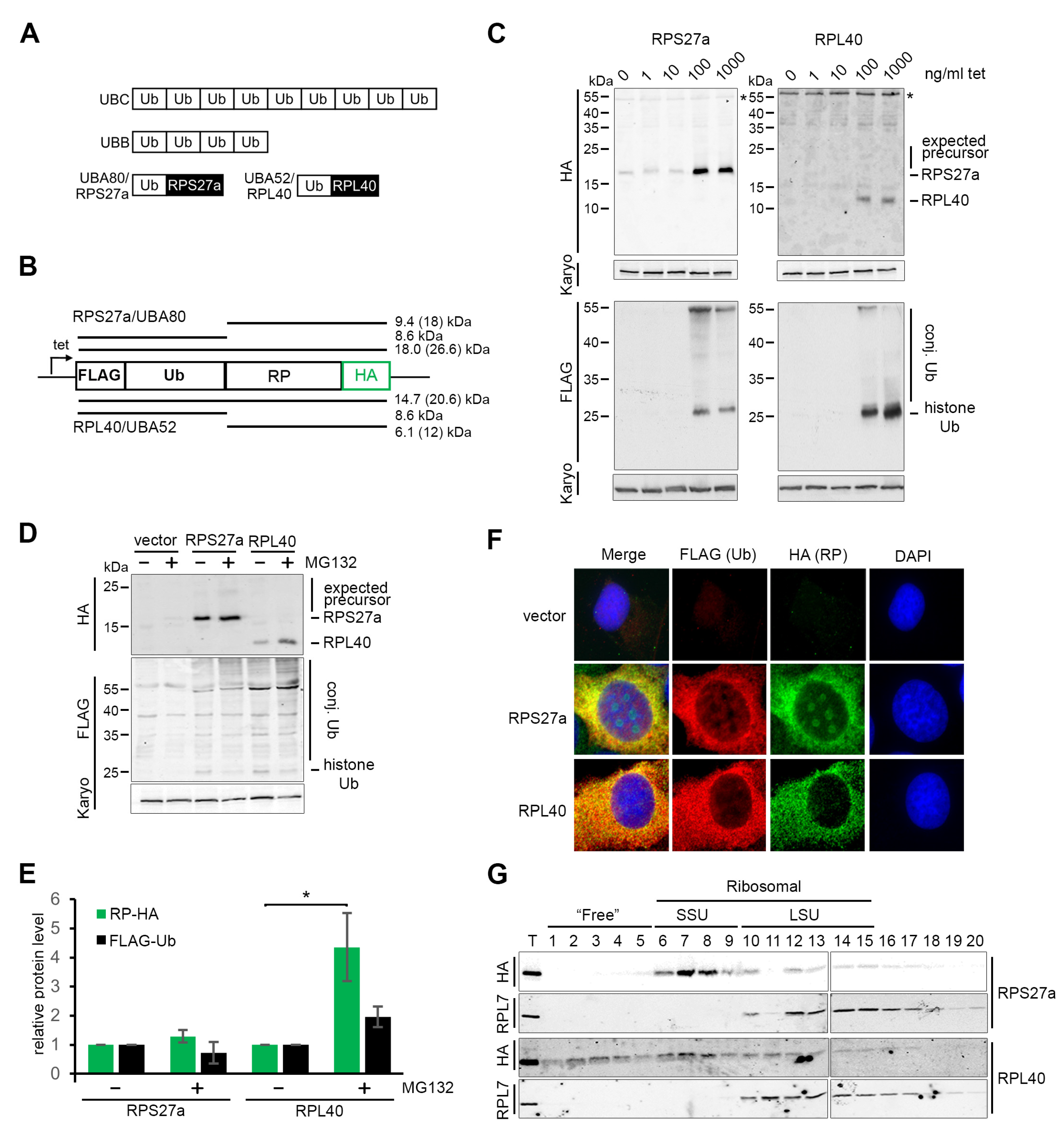
3.1. Both the RPS27a and RPL40 Ubiquitin Fusion Precursors Are Efficiently and Rapidly Cleaved In Vivo
3.2. The Function of RPL40, but Not RPS27a, Is Compromised by the Affinity Tag
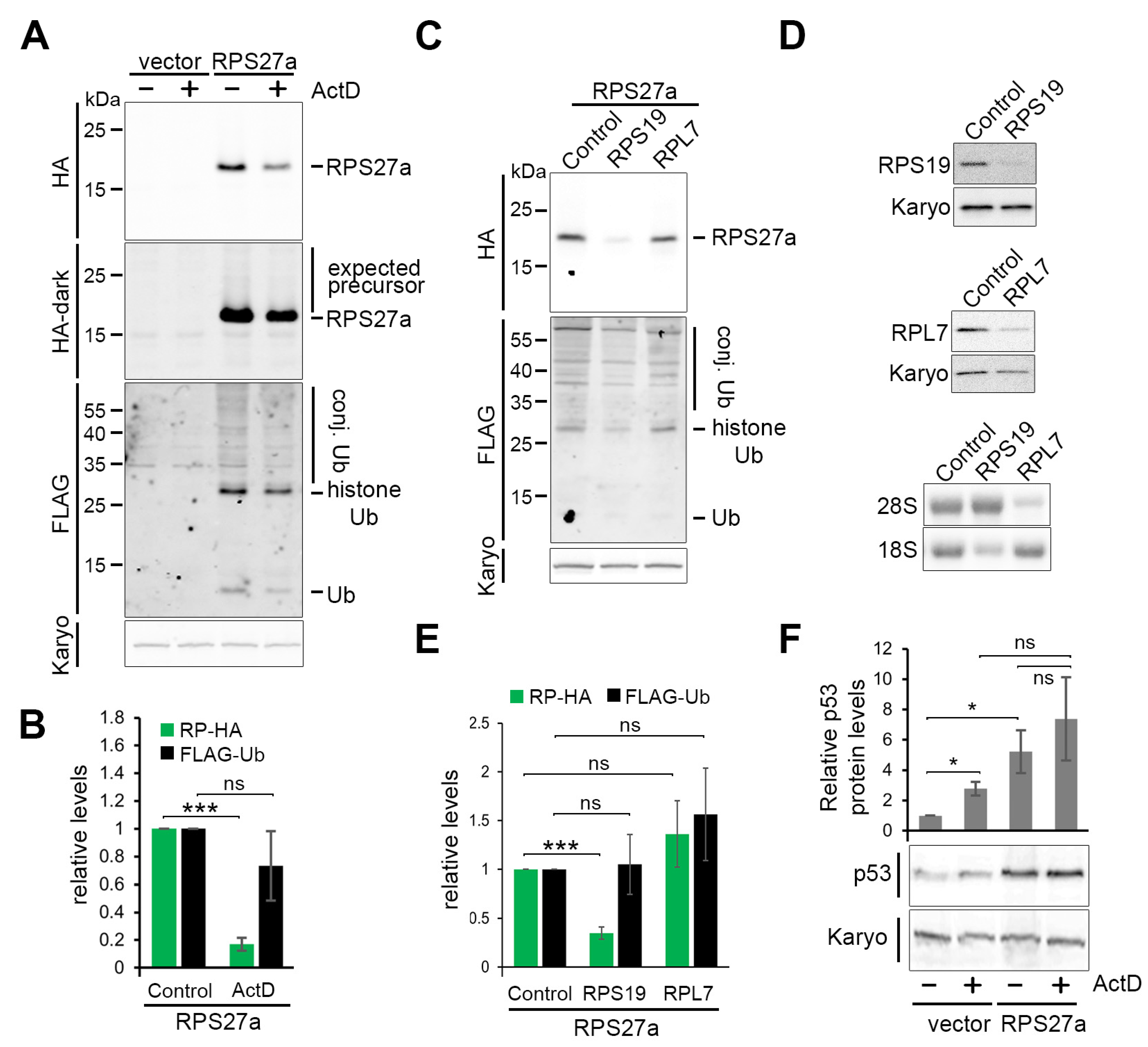
3.3. RPS27a Is Separated from Its Precursor in a Process Independent from Ribosome Biogenesis
3.4. Cells Expressing the Affinity-Tagged Ubiquitin-RPS27a Precursor Exhibit Higher p53 Levels, Which Do Not Change in Response to ActD Treatment
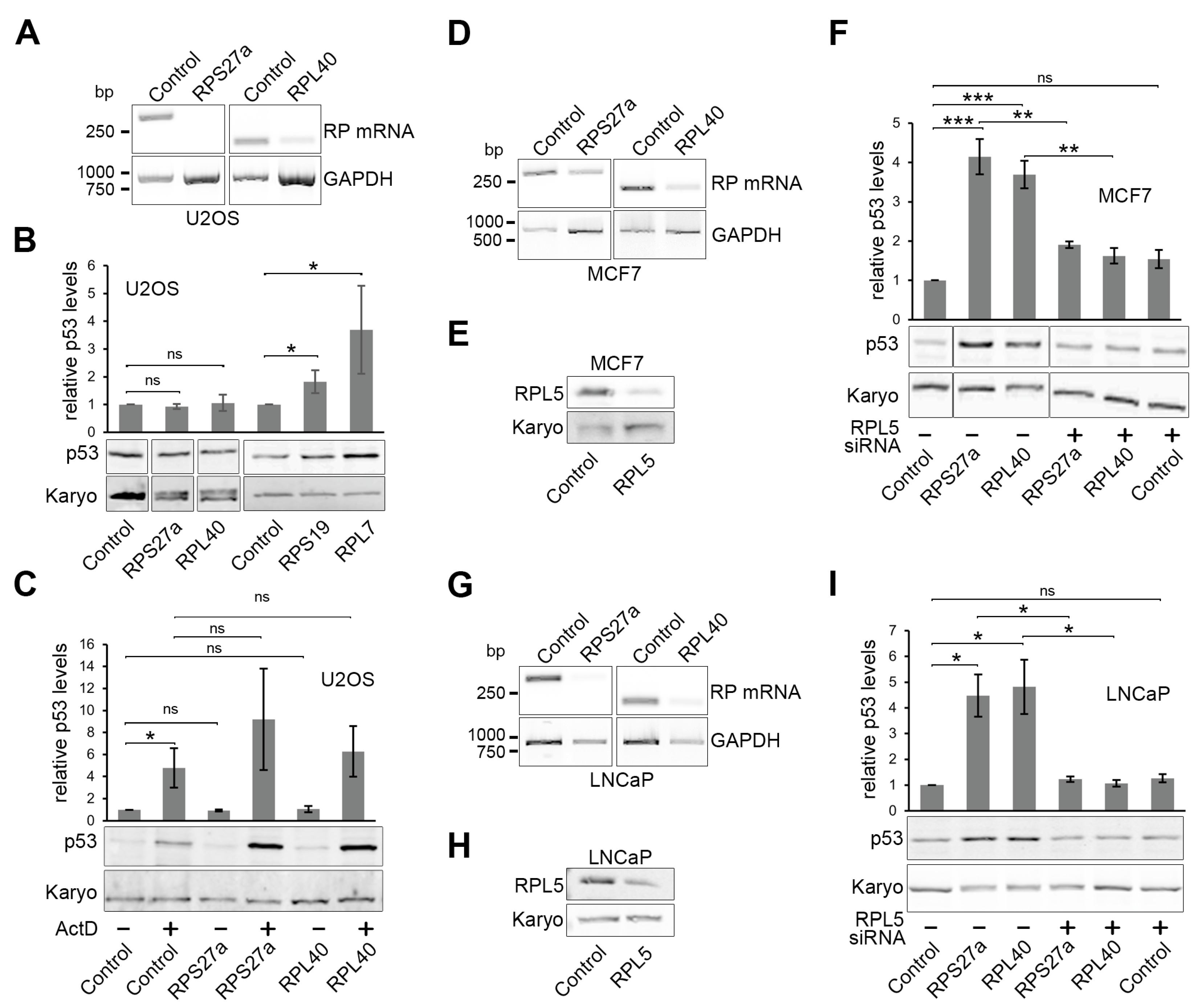
3.5. Knockdown of RPS27a or RPL40 Leads to p53 Stabilisation in MCF7 and LNCaP Cells, but Not in U2OS Cells
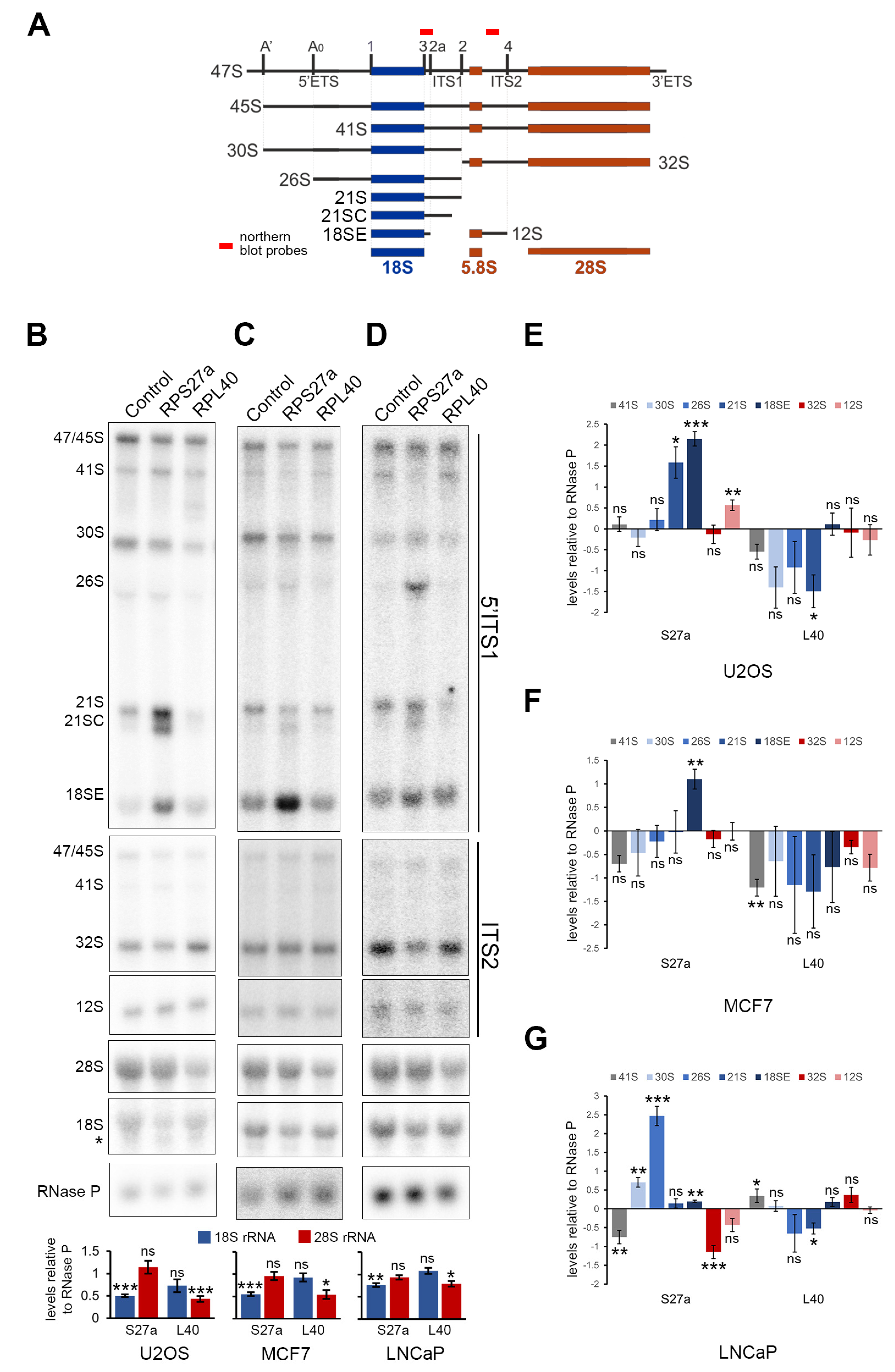
3.6. RPS27a and RPL40 Are Important for Ribosome Production in U2OS, MCF7 and LNCaP Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warner, J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999, 24, 437–440. [Google Scholar] [CrossRef]
- Teng, T.; Thomas, G.; Mercer, C.A. Growth control and ribosomopathies. Curr. Opin. Genet. Dev. 2013, 23, 63–71. [Google Scholar] [CrossRef]
- Lempiainen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Dosil, M. Ribosome biogenesis and cancer: Basic and translational challenges. Curr. Opin. Genet. Dev. 2018, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.F.; Bleichert, F.; Dutca, L.M.; Baserga, S.J. When ribosomes go bad: Diseases of ribosome biogenesis. Mol. Biosyst. 2010, 6, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef]
- Pelava, A.; Schneider, C.; Watkins, N.J. The importance of ribosome production, and the 5S RNP-MDM2 pathway, in health and disease. Biochem. Soc. Trans. 2016, 44, 1086–1090. [Google Scholar] [CrossRef]
- Fumagalli, S.; Thomas, G. The role of p53 in ribosomopathies. Semin. Hematol. 2011, 48, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Bohnsack, M.T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019, 38, e100278. [Google Scholar] [CrossRef]
- Ciganda, M.; Williams, N. Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip. Rev. RNA 2011, 2, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, E.; Plisson-Chastang, C.; Henras, A.K.; Lebaron, S.; Gleizes, P.E.; O’Donohue, M.F.; Romeo, Y.; Henry, Y. Maturation of pre-40S particles in yeast and humans. Wiley Interdiscip. Rev. RNA 2019, 10, e1516. [Google Scholar] [CrossRef]
- Woolford, J.L., Jr.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef]
- Lam, Y.W.; Lamond, A.I.; Mann, M.; Andersen, J.S. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 2007, 17, 749–760. [Google Scholar] [CrossRef]
- Park, C.W.; Ryu, K.Y. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef]
- Lacombe, T.; Garcia-Gomez, J.J.; de la Cruz, J.; Roser, D.; Hurt, E.; Linder, P.; Kressler, D. Linear ubiquitin fusion to Rps31 and its subsequent cleavage are required for the efficient production and functional integrity of 40S ribosomal subunits. Mol. Microbiol. 2009, 72, 69–84. [Google Scholar] [CrossRef]
- Martin-Villanueva, S.; Fernandez-Pevida, A.; Kressler, D.; de la Cruz, J. The Ubiquitin Moiety of Ubi1 Is Required for Productive Expression of Ribosomal Protein eL40 in Saccharomyces cerevisiae. Cells 2019, 8, 850. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Luo, Z.Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef]
- Montellese, C.; van den Heuvel, J.; Ashiono, C.; Dorner, K.; Melnik, A.; Jonas, S.; Zemp, I.; Picotti, P.; Gillet, L.C.; Kutay, U. USP16 counteracts mono-ubiquitination of RPS27a and promotes maturation of the 40S ribosomal subunit. Elife 2020, 9, 1776–1786. [Google Scholar] [CrossRef]
- Kobayashi, M.; Oshima, S.; Maeyashiki, C.; Nibe, Y.; Otsubo, K.; Matsuzawa, Y.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. The ubiquitin hybrid gene UBA52 regulates ubiquitination of ribosome and sustains embryonic development. Sci. Rep. 2016, 6, 36780. [Google Scholar] [CrossRef] [PubMed]
- Martin-Villanueva, S.; Fernandez-Pevida, A.; Fernandez-Fernandez, J.; Kressler, D.; de la Cruz, J. Ubiquitin release from eL40 is required for cytoplasmic maturation and function of 60S ribosomal subunits in Saccharomyces cerevisiae. FEBS J. 2020, 287, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Lee, M.J.; Yu, G.R.; Lee, Z.W.; Bae, J.Y.; Bae, Y.C.; Kang, S.H.; Kim, D.G. Altered dynamics of ubiquitin hybrid proteins during tumor cell apoptosis. Cell Death Dis. 2012, 3, e255. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef]
- Hannan, K.M.; Soo, P.; Wong, M.S.; Lee, J.K.; Hein, N.; Poh, P.; Wysoke, K.D.; Williams, T.D.; Montellese, C.; Smith, L.K.; et al. Nuclear stabilization of p53 requires a functional nucleolar surveillance pathway. Cell Rep. 2022, 41, 111571. [Google Scholar] [CrossRef]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef]
- Dai, M.S.; Zeng, S.X.; Jin, Y.; Sun, X.X.; David, L.; Lu, H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 2004, 24, 7654–7668. [Google Scholar] [CrossRef]
- Sun, X.X.; DeVine, T.; Challagundla, K.B.; Dai, M.S. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J. Biol. Chem. 2011, 286, 22730–22741. [Google Scholar] [CrossRef]
- Yadavilli, S.; Mayo, L.D.; Higgins, M.; Lain, S.; Hegde, V.; Deutsch, W.A. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair 2009, 8, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Leslie, P.; Zhang, Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 2014, 5, 860–871. [Google Scholar] [CrossRef]
- He, X.; Li, Y.; Dai, M.S.; Sun, X.X. Ribosomal protein L4 is a novel regulator of the MDM2-p53 loop. Oncotarget 2016, 7, 16217–16226. [Google Scholar] [CrossRef]
- Bursac, S.; Brdovcak, M.C.; Pfannkuchen, M.; Orsolic, I.; Golomb, L.; Zhu, Y.; Katz, C.; Daftuar, L.; Grabusic, K.; Vukelic, I.; et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. USA 2012, 109, 20467–20472. [Google Scholar] [CrossRef]
- Fumagalli, S.; Ivanenkov, V.V.; Teng, T.; Thomas, G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012, 26, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hou, Z.; Zuo, S.; Zhou, X.; Feng, Y.; Sun, Y.; Yuan, X. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40-MDM2-p53 pathway through binding with UBA52. Cancer Sci. 2019, 110, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, H.; Huang, G.; Bing, Z.; Xu, D.; Liu, J.; Luo, H.; An, X. Loss of RPS27a expression regulates the cell cycle, apoptosis, and proliferation via the RPL11-MDM2-p53 pathway in lung adenocarcinoma cells. J. Exp. Clin. Cancer Res. 2022, 41, 33. [Google Scholar] [CrossRef]
- Al-Hakim, A.K.; Bashkurov, M.; Gingras, A.C.; Durocher, D.; Pelletier, L. Interaction proteomics identify NEURL4 and the HECT E3 ligase HERC2 as novel modulators of centrosome architecture. Mol. Cell. Proteom. 2012, 11, M111.014233. [Google Scholar] [CrossRef]
- Sloan, K.E.; Mattijssen, S.; Lebaron, S.; Tollervey, D.; Pruijn, G.J.; Watkins, N.J. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J. Cell Biol. 2013, 200, 577–588. [Google Scholar] [CrossRef]
- Müller, J.S.; Burns, D.T.; Griffin, H.; Wells, G.R.; Zendah, R.A.; Munro, B.; Schneider, C.; Horvath, R. RNA exosome mutations in pontocerebellar hypoplasia alter ribosome biogenesis and p53 levels. Life Sci. Alliance 2020, 3, e202000678. [Google Scholar] [CrossRef]
- Idol, R.A.; Robledo, S.; Du, H.Y.; Crimmins, D.L.; Wilson, D.B.; Ladenson, J.H.; Bessler, M.; Mason, P.J. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol. Dis. 2007, 39, 35–43. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Weber, K.; Tuschl, T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002, 26, 199–213. [Google Scholar] [CrossRef]
- Turner, A.J.; Knox, A.A.; Prieto, J.L.; McStay, B.; Watkins, N.J. A novel small-subunit processome assembly intermediate that contains the U3 snoRNP, nucleolin, RRP5, and DBP4. Mol. Cell. Biol. 2009, 29, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, Y.; Huang, H.; Li, T.; Ye, L.; Lin, L.; Wei, Y. UBC Mediated by SEPT6 Inhibited the Progression of Prostate Cancer. Mediat. Inflamm. 2021, 2021, 7393029. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Park, S.; Lee, E.K.; Yoo, Y.J. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 2013, 3, 2623. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- O’Donohue, M.F.; Choesmel, V.; Faubladier, M.; Fichant, G.; Gleizes, P.E. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol. 2010, 190, 853–866. [Google Scholar] [CrossRef]
- Fernandez-Pevida, A.; Rodriguez-Galan, O.; Diaz-Quintana, A.; Kressler, D.; de la Cruz, J. Yeast ribosomal protein L40 assembles late into precursor 60 S ribosomes and is required for their cytoplasmic maturation. J. Biol. Chem. 2012, 287, 38390–38407. [Google Scholar] [CrossRef]
- Fernandez-Pevida, A.; Martin-Villanueva, S.; Murat, G.; Lacombe, T.; Kressler, D.; de la Cruz, J. The eukaryote-specific N-terminal extension of ribosomal protein S31 contributes to the assembly and function of 40S ribosomal subunits. Nucleic Acids Res. 2016, 44, 7777–7791. [Google Scholar] [CrossRef]
- Finley, D.; Bartel, B.; Varshavsky, A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 1989, 338, 394–401. [Google Scholar] [CrossRef]
- Ferreira-Cerca, S.; Poll, G.; Gleizes, P.E.; Tschochner, H.; Milkereit, P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol. Cell 2005, 20, 263–275. [Google Scholar] [CrossRef]
- Poll, G.; Braun, T.; Jakovljevic, J.; Neueder, A.; Jakob, S.; Woolford, J.L., Jr.; Tschochner, H.; Milkereit, P. rRNA maturation in yeast cells depleted of large ribosomal subunit proteins. PLoS ONE 2009, 4, e8249. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Bohnsack, M.T.; Schneider, C.; Watkins, N.J. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA 2014, 20, 540–550. [Google Scholar] [CrossRef]
- Wang, M.; Anikin, L.; Pestov, D.G. Two orthogonal cleavages separate subunit RNAs in mouse ribosome biogenesis. Nucleic Acids Res. 2014, 42, 11180–11191. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eastham, M.J.; Pelava, A.; Wells, G.R.; Watkins, N.J.; Schneider, C. RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling. Biomolecules 2023, 13, 898. https://doi.org/10.3390/biom13060898
Eastham MJ, Pelava A, Wells GR, Watkins NJ, Schneider C. RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling. Biomolecules. 2023; 13(6):898. https://doi.org/10.3390/biom13060898
Chicago/Turabian StyleEastham, Matthew John, Andria Pelava, Graeme Raymond Wells, Nicholas James Watkins, and Claudia Schneider. 2023. "RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling" Biomolecules 13, no. 6: 898. https://doi.org/10.3390/biom13060898
APA StyleEastham, M. J., Pelava, A., Wells, G. R., Watkins, N. J., & Schneider, C. (2023). RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling. Biomolecules, 13(6), 898. https://doi.org/10.3390/biom13060898