
The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy



Abstract
:1. Introduction
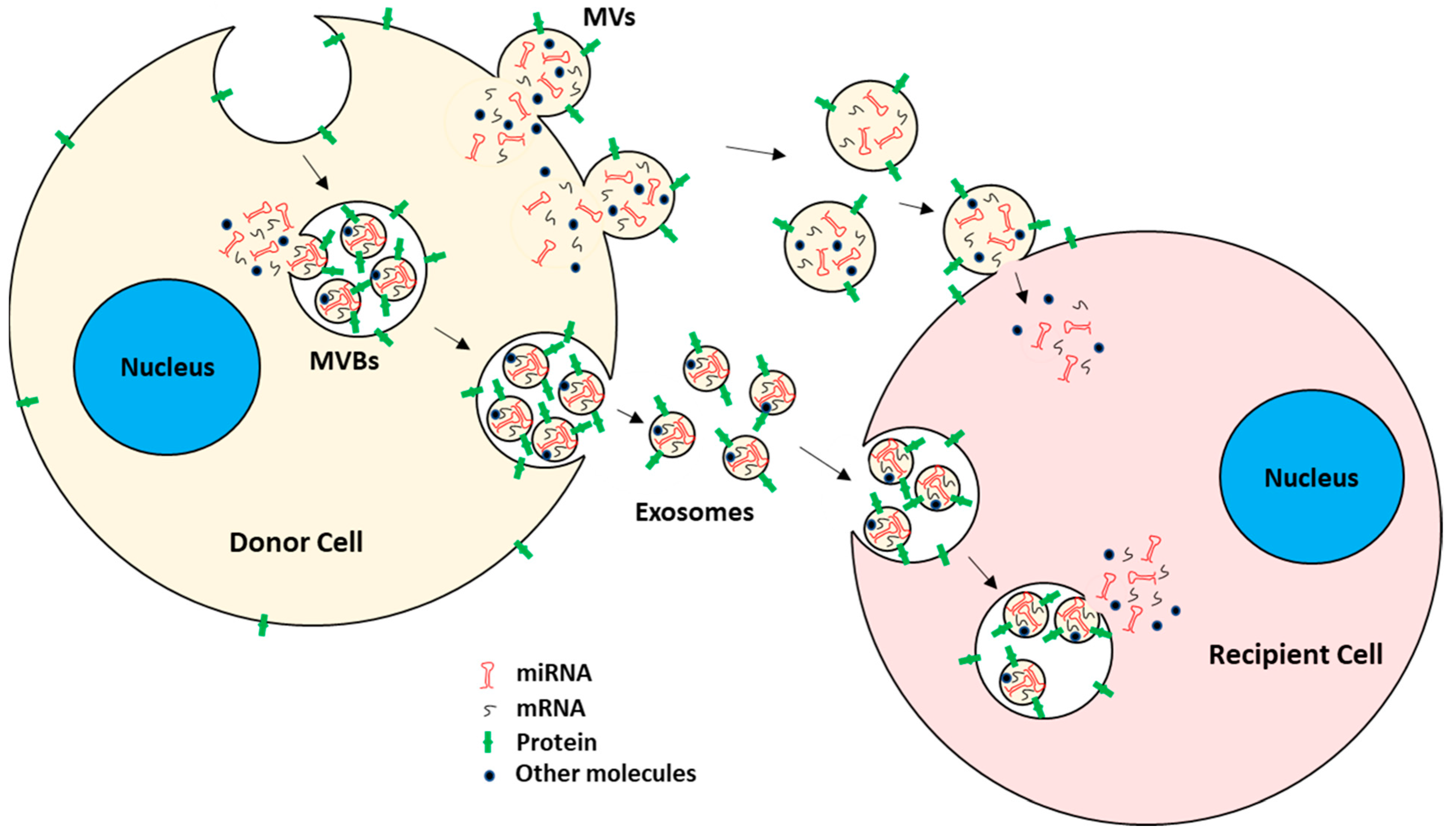
2. Tumor-Derived EVs
3. Hematological Malignancies
4. Hematological Malignancies: The Modern Classification
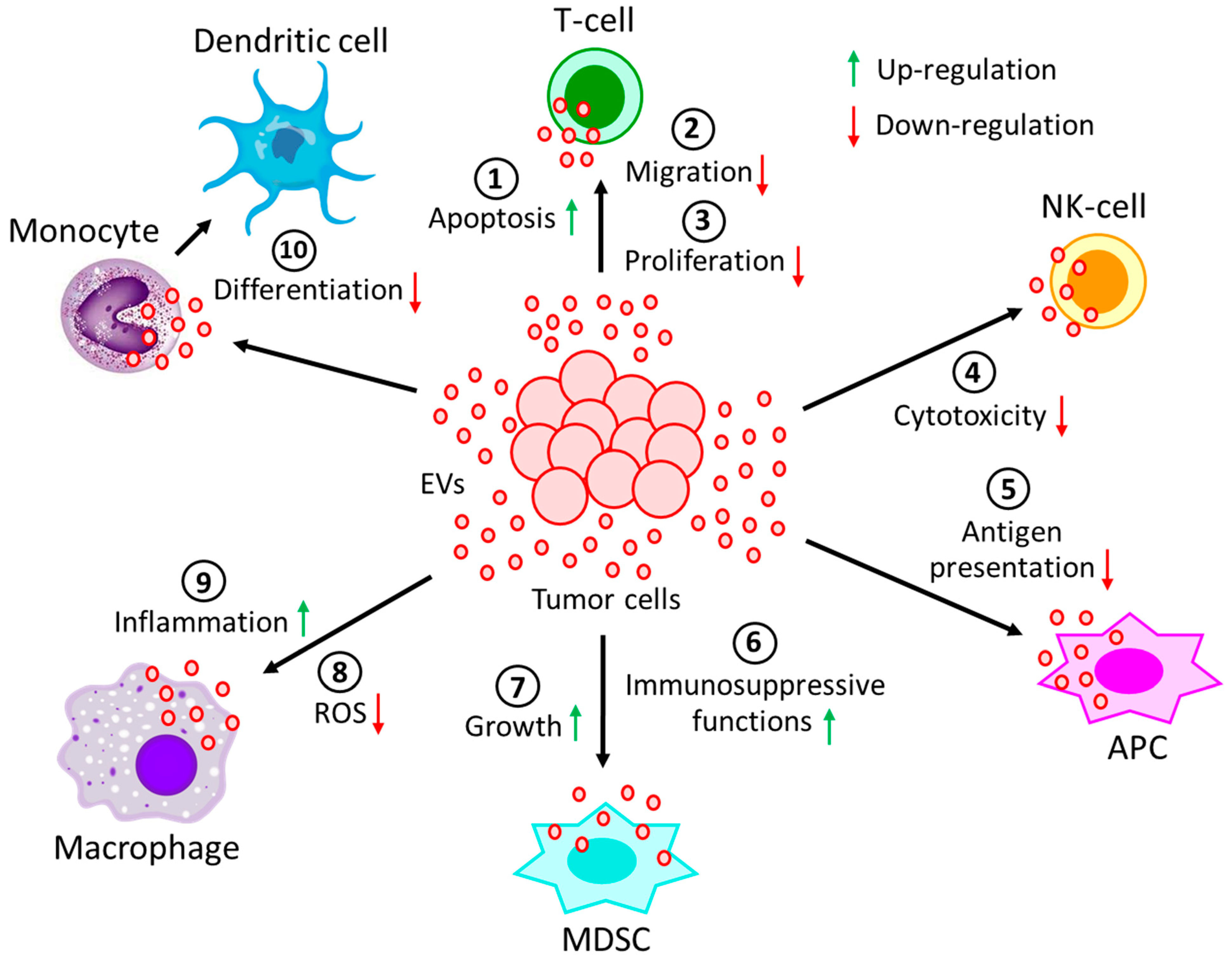
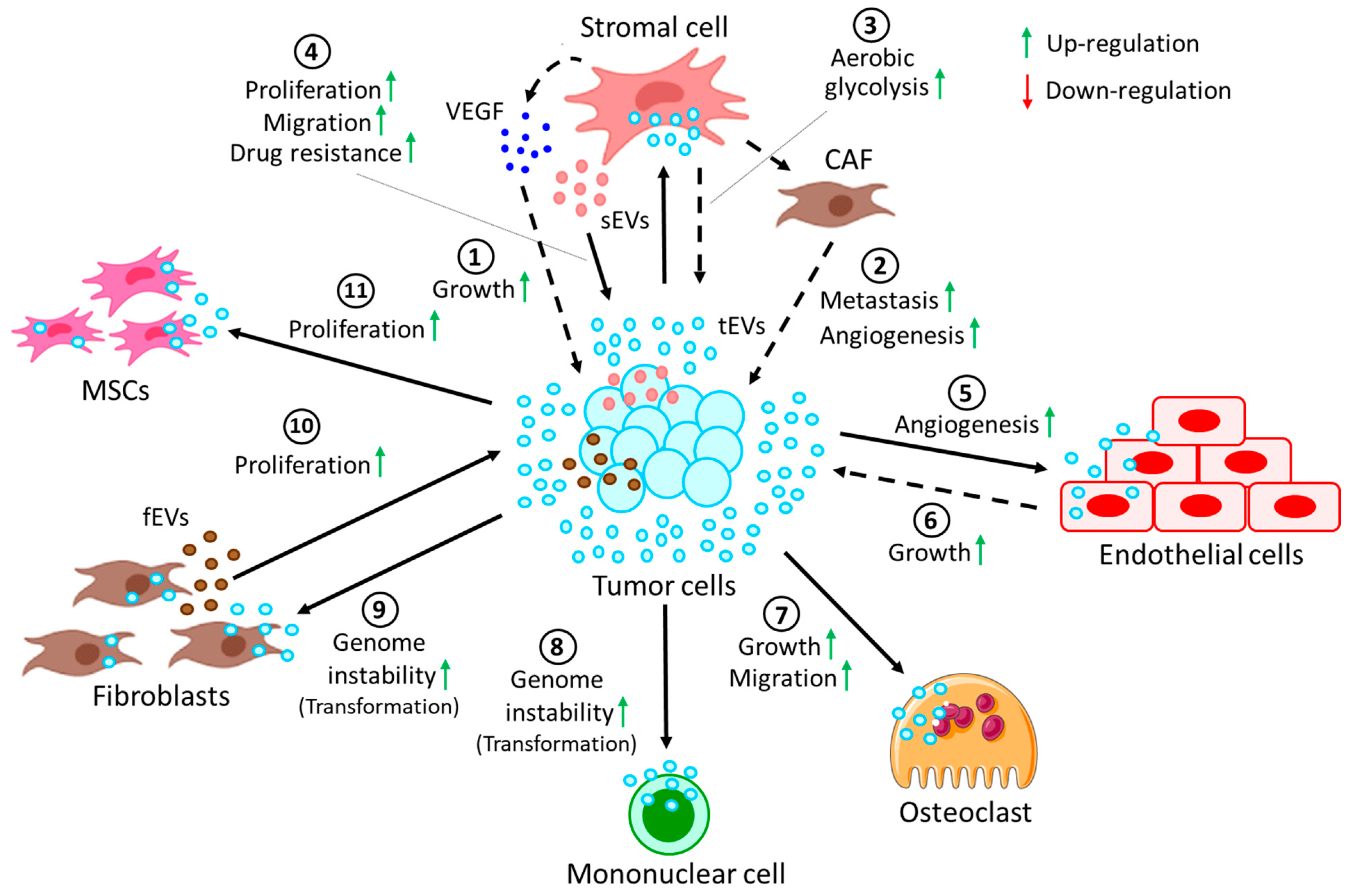
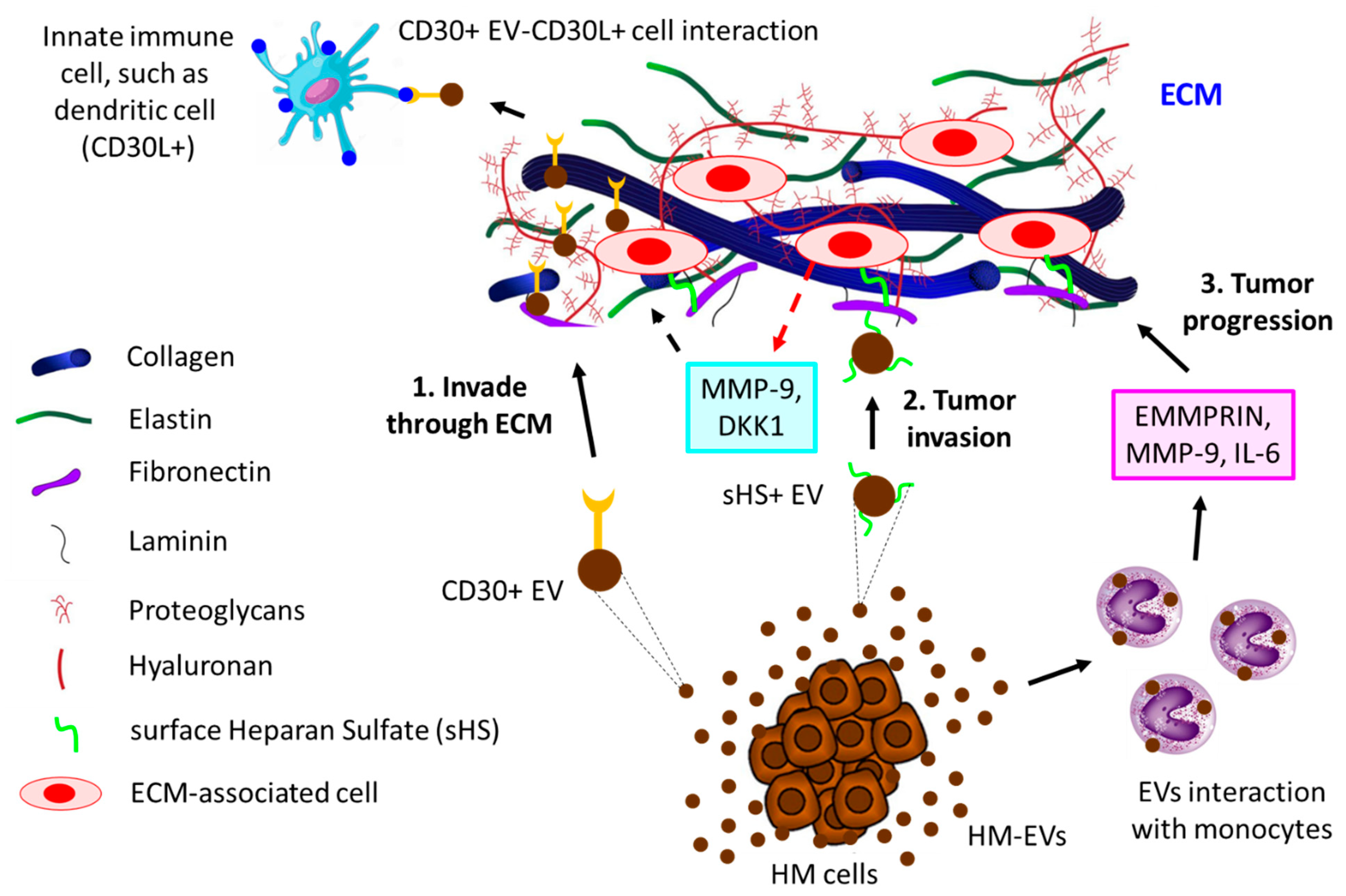
5. EVs in Cell-Cell and Cell-Extracellular Matrix Communication in the TME

{kind=link}
{kind=link}
{kind=link}
{kind=link}
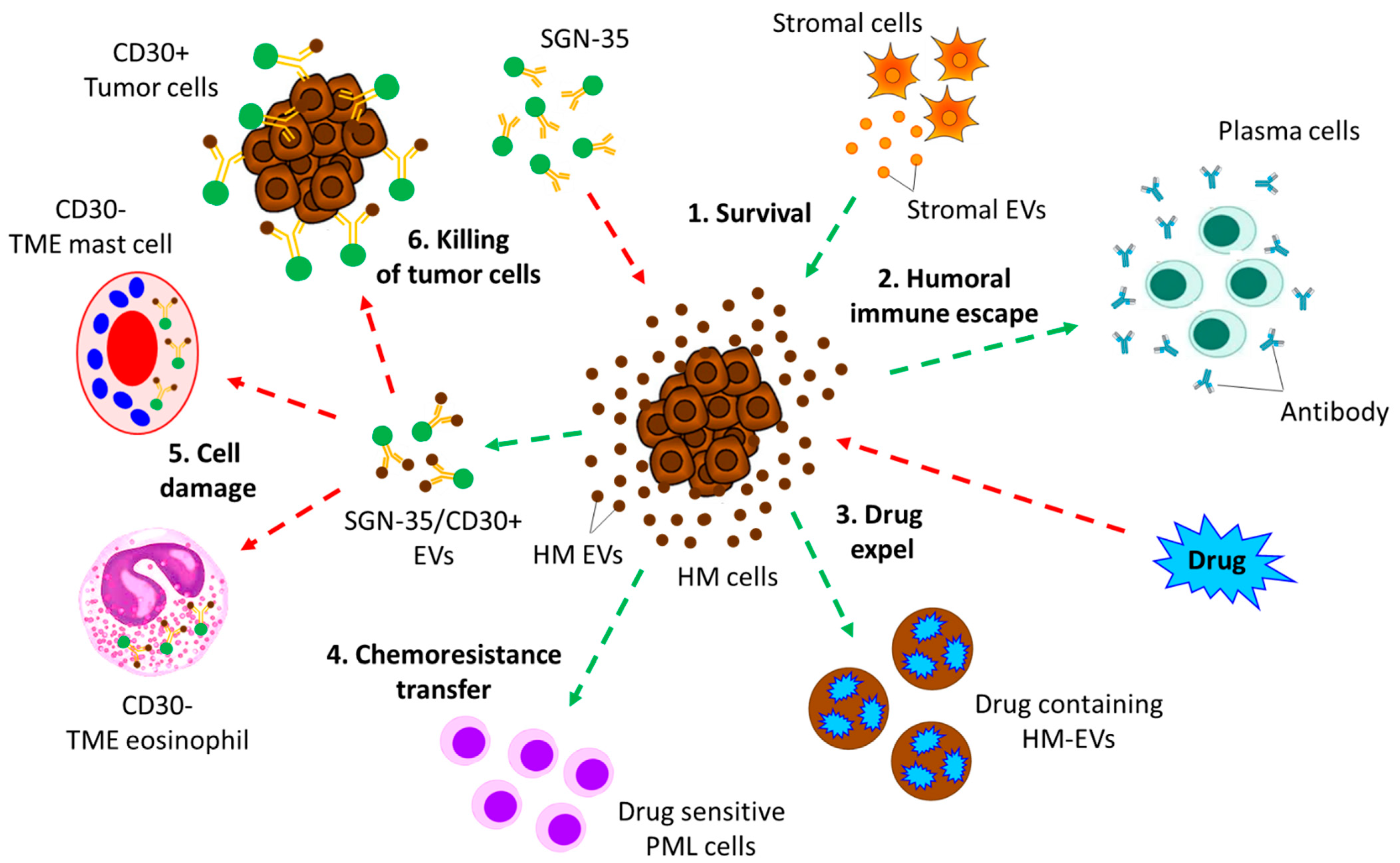{kind=link}
| Originating Cells | Effector Cells | Malignancies | Functions | References |
|---|---|---|---|---|
| Leukemic cells | CD8(+)T-cells | Solid tumors or AML | Down-regulates CD3ζ and JAK3 expression and promotes Fas/FasL-mediated T-cell apoptosis | [108] |
| Leukemic cells | CD4(+)T-cells | CLL | Down-regulates CD69 expression via miR-363 transfer and affects effector T-cell migration | [109,110,111] |
| Lymphoma cells | T-cells | DLBCL | Induces PD-1 expression in T-cells and enhances T-cell apoptosis | [112] |
| Lymphoma cells | T-cells | BCL | Carries CD39 and CD73 and hydrolyzes ATP to generate adenosine to inhibit T-cell activity and proliferation | [114] |
| Lymphoma cells | NK-cells, APCs | BCL, TCL | Carries MHC, APO2L, FASL, TCR, and NKG2D and inhibits NK-cells cytotoxicity and antigen processing of APCs | [115,116,117] |
| Non-leukemic cells | NK-cells | CLL | Carries BAG6 and activates NK-cells, but activated NK-cells are eliminated by lymphocytes | [118] |
| Myeloma cells | MDSCs, | MM | Induces growth and immunosuppressive activity | [119,120] |
| NK-cells, | Reduces NK-cells’ cytotoxicity | [121] | ||
| Immune cells | Carries ectoenzyme, CD38, which converts nucleotides into adenosine to suppress immune system | [122] | ||
| Leukemic cells | NK-cells | AML | EVs’-bound TGFβ1 reduces NK-cells’ cytotoxicity | [123] |
| Lymphoma cells | Monocytes | - | Releases TNF-α, IL-1β, and IL-6 and prevents monocyte differentiation into dendritic cells | [124] |
| Lymphoma cells | Macrophages | DLBCL | Transfers MyD88 and stimulates pro-inflammatory NF-κB signaling pathway | [125] |
| Leukemic cells | Macrophages | CML | Polarizes macrophages to M2-phenotype to induce TNF-α and IL-10 expression and down-regulates NO and ROS generation | [126] |
| Tumor cells | Neutrophils | CAT | Promotes NET formation, reduced generation of suppressor cells | [127,128,129] |
| Lymphoma cells | MDSCs | Lymphoma | Carries HSP72 and promotes suppressive functions | [124] |
| Originating Cells | Effector Cells | Malignancies | Functions | References |
|---|---|---|---|---|
| Tumor cells | Stromal cells | CLL | Tumor-derived EVs induce stromal cells to release VEGF to promote tumor survival | [139] |
| Tumor cells | Stromal cells | CLL | Tumor-derived EVs convert stromal cells into CAFs, thereby promoting metastasis and angiogenesis | [140,141] |
| Stromal cells | Cancer cells | ALL | Stromal cell-derived EVs induce GAL3 expression in cancer cells, hence inducing drug resistance | [142] |
| Tumor cells | Stromal cells | ALL | Switch oxidative phosphorylation to aerobic glycolysis in favor of cancer progression | [143] |
| Stromal cells | Tumor cells | MM | Stromal cells from MM induce proliferation, migration, and survival of tumor cells | [144] |
| Tumor cells | Endothelial cells | MM | Tumor cell-derived EVs in hypoxic conditions affect miR-135b, targeting HIF-1 pathway to promote angiogenesis | [145] |
| Tumor cells | Endothelial cells | MM | Tumor cell-secreted EVs activate endothelial STAT3 pathway, which promotes angiogenesis and tumor growth | [146] |
| Tumor cells | Osteoclasts | MM | MM-derived EVs support osteoclast growth and migration | [147] |
| Fibroblasts | Tumor cells | MM | EVs carry clBcl-xL, which helps in EV uptake by the tumor cells to promote tumor proliferation | [148] |
| Tumor cells | MSCs | ATL | Tumor cell-derived EVs transfer miR-155 and miR-21 to the MSCs, thereby inducing MSC proliferation | [149] |
| Leukemic CD34+ cells | MSCs | AML | AML CD34+ cell-derived EVs reduce further development of CD34+ cells from MSCs via miR-7977 | [150] |
| MSCs | CD34+ cells | MPN | EVs carry miR-155 from MSCs, increasing granulocyte CFU numbers in neoplastic CD34+ cells | [151] |
| Leukemic cells | MSCs | CML | CML-derived EVs induce IL-8 release from MSCs, thereby promoting CML survival | [152] |
| BCR-ABL + tumor cells | Mononuclear | CML | Induce genome instability, leading to malignant transformation of cells | [153] |
| Tumor cells | Fibroblasts | TCL, CML | Transfer of hTERT mRNA via the EVs results in induced hTERT expression in the fibroblasts, leading to genome instability | [154,155] |
6. EVs: The Biomarker of Hematological Malignancies: Contribution in Drug Resistance
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EV | extracellular vesicle |
| MV | microvesicle |
| SNARE | soluble N-ethylmaleimide-sensitive factor activating protein receptor |
| MVB | multivesicular body |
| ESCRT | endosomal sorting complexes required for transport |
| TSG101 | tumor susceptibility gene 101 |
| HSC70 | heat shock cognate 70-kDa protein |
| HSP90β | heat shock protein 90 beta |
| CD | cluster of differentiation |
| GRP | Golgi and endoplasmic reticulum associated protein |
| CSE1L | chromosome segregation 1 like |
| TrpC5 | transient receptor potential cation channel subfamily C member 5 |
| EMT | epithelial to mesenchymal transition |
| JAK | Janus kinase |
| PD-L1 | programmed death ligand 1 |
| PD1 | programmed cell death protein 1 |
| CLL | chronic lymphocytic leukemia |
| DLBCL | diffuse large B cell lymphoma |
| BCL | B-cell lymphoma |
| ATP | adenosine triphosphate |
| MHC | major histocompatibility complex |
| APO2L | Apo2 ligand |
| TCR | T-cell receptor |
| NKG2D | natural-killer group-2 member-D |
| APC | antigen presenting cell |
| NKp30 | natural killer protein 30 |
| BAG6 | Bcl2-associated athanogene cochaperone 6 |
| MM | multiple myeloma |
| MDSC | myeloid-derived suppressor cell |
| TGFβ1 | transforming growth factor beta 1 |
| AML | acute myeloid leukemia |
| NK | natural killer |
| TNF-α | tumor necrosis factor α |
| IL | interleukin |
| MyD88 | myeloid differentiation primary response 88 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| TLR | Toll-like receptor |
| IL-1R | interleukin-1 receptor |
| CML | chronic myelogenous leukemia |
| NO | nitric oxide |
| ROS | reactive oxygen species |
| TAM | tumor-associated macrophage |
| NET | neutrophil extracellular trap |
| VEGF | vascular endothelial growth factor |
| CAF | cancer-associated fibroblast |
| ALL | acute lymphoblastic leukemia |
| GAL3 | galectin 3 |
| HIF | hypoxia inducible factor |
| STAT | signal transducer and activator of transcription |
| OC | osteoclast |
| clBcl-xL | cleaved Bcl-xL |
| MSC | mesenchymal stem cell |
| ATL | adult T-cell leukemia/lymphoma |
| MPN | myeloproliferative neoplasm |
| CFU | colony forming unit |
| BCR | B-cell receptor |
| hTERT | human telomerase reverse transcriptase |
| TCL | T-cell lymphoma |
| sHS | surface heparan sulfate |
| MAPK | mitogen-activated protein kinase |
| ERK | extracellular signal-regulated kinase |
| MMP | matrix metalloproteinase |
| EMMPRIN | extracellular matrix metalloproteinase inducer |
| NOTCH | neurogenic locus notch homolog protein |
| ABC | ATP binding cassette |
| GLOBOCAN | Global Cancer Observatory |
| WHO | World Health Organization |
| ABL1 | Abelson murine leukemia viral oncogene homolog 1 |
| PV | polycythemia vera |
| ET | essential thrombocytopenia |
| PF | primary myelofibrosis |
| CNL | chronic neutrophilic leukemia |
| CEL | chronic eosinophilic leukemia |
| MDS | myelodysplastic neoplasms/syndrome |
| BMB | bone marrow blasts |
| PB | peripheral blasts |
| SF3B1 | splicing factor 3B subunit 1 |
| TP53 | tumor protein 53 |
| IB | increased blasts |
| NOS | not otherwise specified |
| DS | Down syndrome |
| SLL | small lymphocytic lymphoma |
| LPL | lymphoplasmacytic lymphoma |
| MGUS | monoclonal gammopathy of undetermined significance |
| IgM | immunoglobulin M |
| MZL | marginal zone lymphoma |
| FL | follicular lymphoma |
| MCL | mantle cell lymphoma |
| BL | Burkitt lymphoma |
| HL | Hodgkin lymphoma |
| cHL | classic HL |
| NLPHL | nodular lymphocyte-predominant HL |
| PTCL | peripheral T-cell lymphoma |
| ALCL | anaplastic large cell lymphoma |
| ALK | anaplastic lymphoma kinase |
| AITL | angioimmunoblastic T-cell lymphoma |
| EBV | Epstein–Barr virus |
| HTLV | human T-lymphotropic virus |
| TLGL | T-cell large granular lymphocyte leukemia |
| NKLGL | NK-cell large granular lymphocyte leukemia |
| TK | tyrosine kinase |
| HE | hyper eosinophilia |
| AEC | absolute eosinophil count |
| HES | hypereosinophilic syndrome |
| PDGFR | platelet-derived growth factor receptor |
| FGFR1 | fibroblast growth factor receptor 1 |
| ETV6 | ETS (erythroblast transformation specific) variant transcription factor 6 |
| FLT3 | FMS-like tyrosine kinase 3 |
| CM | cutaneous mastocytosis |
| SM | systemic mastocytosis |
| MPAL | mixed phenotype acute leukemia |
| piRNA | Piwi-interacting RNA |
| VCAM-1 | vascular cell adhesion molecule 1 |
| CXCL | chemokine (C-X-C motif) ligand |
| PML | progressive multifocal leukoencephalopathy |
| RAR-α | retinoic acid receptor α |
| sPLA2-X | soluble phospholipase A2 of group X |
| TNFAIP3 | tumor necrosis factor alpha-induced protein 3 |
| ITD | internal tandem duplication |
| IGF-IR | insulin-like growth factor 1 (IGF-1) receptor |
| NPM1 | nucleophosmin gene 1 |
| HLA | human leukocyte antigen |
| HRS | Hodgkin and Reed–Sternberg |
| WM | Waldenstrom macroglobulinemia |
| BMSC | bone-marrow stromal cell |
| HM | hematological malignant |
References
- Combarnous, Y.; Nguyen, T.M.D. Cell Communications among Microorganisms, Plants, and Animals: Origin, Evolution, and Interplays. Int. J. Mol. Sci. 2020, 21, 8052. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wang, R. Exploring the mechanisms of cell reprogramming and transdifferentiation via intercellular communication. Phys. Rev. E 2020, 102, 012406. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Prasad, R.; Roy, S.; Mukherjee, A.; Sen, P. The Protease Activated Receptor2 Promotes Rab5a Mediated Generation of Pro-metastatic Microvesicles. Sci. Rep. 2018, 8, 7357. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Prasad, R.; Singh, A.; Bhattacharya, A.; Roy, A.; Mallik, S.; Mukherjee, A.; Sen, P. Protease-activated receptor 2 promotes actomyosin dependent transforming microvesicles generation from human breast cancer. Mol. Carcinog. 2018, 57, 1707–1722. [Google Scholar] [CrossRef]
- Das, K.; Paul, S.; Singh, A.; Ghosh, A.; Roy, A.; Ansari, S.A.; Prasad, R.; Mukherjee, A.; Sen, P. Triple-negative breast cancer-derived microvesicles transfer microRNA221 to the recipient cells and thereby promote epithelial-to-mesenchymal transition. J. Biol. Chem. 2019, 294, 13681–13696. [Google Scholar] [CrossRef]
- Das, K.; Keshava, S.; Ansari, S.A.; Kondreddy, V.; Esmon, C.T.; Griffin, J.H.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa induces extracellular vesicles from the endothelium: A potential mechanism for its hemostatic effect. Blood 2021, 137, 3428–3442. [Google Scholar] [CrossRef]
- Das, K.; Keshava, S.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa suppresses inflammation and barrier disruption through the release of EEVs and transfer of microRNA 10a. Blood 2022, 139, 118–133. [Google Scholar] [CrossRef]
- Das, K.; Pendurthi, U.R.; Manco-Johnson, M.; Martin, E.J.; Brophy, D.F.; Rao, L.V.M. Factor VIIa treatment increases circulating extracellular vesicles in hemophilia patients: Implications for the therapeutic hemostatic effect of FVIIa. J. Thromb. Haemost. 2022, 20, 1928–1933. [Google Scholar] [CrossRef]
- Das, K.; Rao, L.V.M. The Role of microRNAs in Inflammation. Int. J. Mol. Sci. 2022, 23, 15479. [Google Scholar] [CrossRef]
- Gonzalez Fernandez, J.; Moncayo Arlandi, J.; Ochando, A.; Simon, C.; Vilella, F. The role of extracellular vesicles in intercellular communication in human reproduction. Clin. Sci. 2023, 137, 281–301. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Yin, Y.; Jia, X.; Mao, L. Mechanism of cargo sorting into small extracellular vesicles. Bioengineered 2021, 12, 8186–8201. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Neven, K.Y.; Nawrot, T.S.; Bollati, V. Extracellular Vesicles: How the External and Internal Environment Can Shape Cell-To-Cell Communication. Curr. Environ. Health Rep. 2017, 4, 30–37. [Google Scholar] [CrossRef]
- Muraoka, S.; Hirano, M.; Isoyama, J.; Ishida, M.; Tomonaga, T.; Adachi, J. Automated Proteomics Sample Preparation of Phosphatidylserine-Positive Extracellular Vesicles from Human Body Fluids. ACS Omega 2022, 7, 41472–41479. [Google Scholar] [CrossRef]
- Pulliero, A.; Pergoli, L.; La Maestra, S.; Micale, R.T.; Camoirano, A.; Bollati, V.; Izzotti, A.; De Flora, S. Extracellular vesicles in biological fluids. A biomarker of exposure to cigarette smoke and treatment with chemopreventive drugs. J. Prev. Med. Hyg. 2019, 60, E327–E336. [Google Scholar] [CrossRef]
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. 2013, 46, 824–830. [Google Scholar] [CrossRef]
- Liu, Y.J.; Wang, C. A review of the regulatory mechanisms of extracellular vesicles-mediated intercellular communication. Cell Commun. Signal. 2023, 21, 77. [Google Scholar] [CrossRef]
- Clancy, J.W.; Schmidtmann, M.; D’Souza-Schorey, C. The ins and outs of microvesicles. FASEB Bioadv. 2021, 3, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Reinisch, K.; Ferro-Novick, S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev. Cell 2007, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Plawinski, L.; Cras, A.; Hernandez Lopez, J.R.; de la Pena, A.; Van der Heyden, A.; Belle, C.; Toti, F.; Angles-Cano, E. Distinguishing Plasmin-Generating Microvesicles: Tiny Messengers Involved in Fibrinolysis and Proteolysis. Int. J. Mol. Sci. 2023, 24, 1571. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Zoller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Di Vizio, D.; Morello, M.; Dudley, A.C.; Schow, P.W.; Adam, R.M.; Morley, S.; Mulholland, D.; Rotinen, M.; Hager, M.H.; Insabato, L.; et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol. 2012, 181, 1573–1584. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef]
- Morello, M.; Minciacchi, V.R.; de Candia, P.; Yang, J.; Posadas, E.; Kim, H.; Griffiths, D.; Bhowmick, N.; Chung, L.W.; Gandellini, P.; et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 2013, 12, 3526–3536. [Google Scholar] [CrossRef]
- Rech, J.; Getinger-Panek, A.; Gałka, S.; Bednarek, I. Origin and Composition of Exosomes as Crucial Factors in Designing Drug Delivery Systems. Appl. Sci. 2022, 12, 12259. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Kim, Y.B.; Lee, G.B.; Moon, M.H. Size Separation of Exosomes and Microvesicles Using Flow Field-Flow Fractionation/Multiangle Light Scattering and Lipidomic Comparison. Anal. Chem. 2022, 94, 8958–8965. [Google Scholar] [CrossRef]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Babst, M.; Katzmann, D.J.; Estepa-Sabal, E.J.; Meerloo, T.; Emr, S.D. Escrt-III: An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell 2002, 3, 271–282. [Google Scholar] [CrossRef]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shin, K.J.; Jang, H.J.; Ryu, J.S.; Lee, C.Y.; Yoon, J.H.; Seo, J.K.; Park, S.; Lee, S.; Je, A.R.; et al. GPR143 controls ESCRT-dependent exosome biogenesis and promotes cancer metastasis. Dev. Cell 2023, 58, 320–334.e328. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Ji, H.; Mathivanan, S.; Scott, A.M.; Simpson, R.J. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods 2012, 56, 293–304. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Geminard, C.; De Gassart, A.; Blanc, L.; Vidal, M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic 2004, 5, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Buschow, S.I.; van Balkom, B.W.; Aalberts, M.; Heck, A.J.; Wauben, M.; Stoorvogel, W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 2010, 88, 851–856. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Kalluri, R.; McAndrews, K.M. The role of extracellular vesicles in cancer. Cell 2023, 186, 1610–1626. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Wang, Y.; Khan, H.M.; Zhou, C.; Liao, X.; Tang, P.; Song, P.; Gui, X.; Li, H.; Chen, Z.; Liu, S.; et al. Apoptotic cell-derived micro/nanosized extracellular vesicles in tissue regeneration. Nanotechnol. Rev. 2022, 11, 957–972. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023. [Google Scholar] [CrossRef]
- Bano, R.; Ahmad, F.; Mohsin, M. A perspective on the isolation and characterization of extracellular vesicles from different biofluids. RSC Adv. 2021, 11, 19598–19615. [Google Scholar] [CrossRef]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef]
- Zhang, M.; Lin, Y.; Chen, R.; Yu, H.; Li, Y.; Chen, M.; Dou, C.; Yin, P.; Zhang, L.; Tang, P. Ghost messages: Cell death signals spread. Cell Commun. Signal. 2023, 21, 6. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Maisano, D.; Mimmi, S.; Dattilo, V.; Marino, F.; Gentile, M.; Vecchio, E.; Fiume, G.; Nistico, N.; Aloisio, A.; de Santo, M.P.; et al. A novel phage display based platform for exosome diversity characterization. Nanoscale 2022, 14, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, H.; Mao, J.; Cao, H.; Tao, Y.; Zhao, G.; Zhang, Z.; Zhang, N.; Liu, Z.; Zhang, J.; et al. Exosome-based nanoimmunotherapy targeting TAMs, a promising strategy for glioma. Cell Death Dis. 2023, 14, 235. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, K.S.; Francescone, R.; Franco-Barraza, J.; Gardiner, J.C.; Vendramini-Costa, D.B.; Luong, T.; Pourmandi, N.; Andren, A.; Kurimchak, A.; Ogier, C.; et al. NetrinG1(+) cancer-associated fibroblasts generate unique extracellular vesicles that support the survival of pancreatic cancer cells under nutritional stress. Cancer Res. Commun. 2022, 2, 1017–1036. [Google Scholar] [CrossRef] [PubMed]
- Fathi, M.; Martinez-Paniagua, M.; Rezvan, A.; Montalvo, M.J.; Mohanty, V.; Chen, K.; Mani, S.A.; Varadarajan, N. Identifying signatures of EV secretion in metastatic breast cancer through functional single-cell profiling. iScience 2023, 26, 106482. [Google Scholar] [CrossRef]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Jelinek, D.F. Multiple myeloma dell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget 2014, 5, 5686–5699. [Google Scholar] [CrossRef]
- McMasters, A.; McMasters, K.M.; Hao, H. Exosome to Promote Cancer Progression via Its Bioactive Cargoes. Arch. Cancer Biol. Ther. 2021, 2, 29–34. [Google Scholar]
- Barlin, M.; Erdmann-Gilmore, P.; Mudd, J.L.; Zhang, Q.; Seymour, R.W.; Guo, Z.; Miessner, J.R.; Goedegebuure, S.P.; Bi, Y.; Osorio, O.A.; et al. Proteins in Tumor-Derived Plasma Extracellular Vesicles Indicate Tumor Origin. Mol. Cell. Proteom. 2023, 22, 100476. [Google Scholar] [CrossRef]
- Liao, C.F.; Lin, S.H.; Chen, H.C.; Tai, C.J.; Chang, C.C.; Li, L.T.; Yeh, C.M.; Yeh, K.T.; Chen, Y.C.; Hsu, T.H.; et al. CSE1L, a novel microvesicle membrane protein, mediates Ras-triggered microvesicle generation and metastasis of tumor cells. Mol. Med. 2012, 18, 1269–1280. [Google Scholar] [CrossRef]
- Dong, Y.; Pan, Q.; Jiang, L.; Chen, Z.; Zhang, F.; Liu, Y.; Xing, H.; Shi, M.; Li, J.; Li, X.; et al. Tumor endothelial expression of P-glycoprotein upon microvesicular transfer of TrpC5 derived from adriamycin-resistant breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 85–90. [Google Scholar] [CrossRef]
- Jiang, J.; Li, J.; Zhou, X.; Zhao, X.; Huang, B.; Qin, Y. Exosomes Regulate the Epithelial-Mesenchymal Transition in Cancer. Front. Oncol. 2022, 12, 864980. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Pukhalskaia, T.V.; Rizvanov, A.A.; Solovyeva, V.V. Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells. Bioengineering 2022, 9, 245. [Google Scholar] [CrossRef]
- Robado de Lope, L.; Sanchez-Herrero, E.; Serna-Blasco, R.; Provencio, M.; Romero, A. Cancer as an infective disease: The role of EVs in tumorigenesis. Mol. Oncol. 2023, 17, 390–406. [Google Scholar] [CrossRef]
- Salem, A.E.; Shah, H.R.; Covington, M.F.; Koppula, B.R.; Fine, G.C.; Wiggins, R.H.; Hoffman, J.M.; Morton, K.A. PET-CT in Clinical Adult Oncology: I. Hematologic Malignancies. Cancers 2022, 14, 5941. [Google Scholar] [CrossRef]
- Sochacka-Cwikla, A.; Maczynski, M.; Regiec, A. FDA-Approved Drugs for Hematological Malignancies-The Last Decade Review. Cancers 2021, 14, 87. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, Y.; Yu, K.; Chen, S.; Wang, W.; Gale, R.P.; Liu, Z.X.; Liang, Y. Changing causes of death in persons with haematological cancers 1975–2016. Leukemia 2022, 36, 1850–1860. [Google Scholar] [CrossRef]
- Wedding, U. Palliative care of patients with haematological malignancies: Strategies to overcome difficulties via integrated care. Lancet Healthy Longev. 2021, 2, e746–e753. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Vijenthira, A.; Gong, I.Y.; Fox, T.A.; Booth, S.; Cook, G.; Fattizzo, B.; Martin-Moro, F.; Razanamahery, J.; Riches, J.C.; Zwicker, J.; et al. Outcomes of patients with hematologic malignancies and COVID-19: A systematic review and meta-analysis of 3377 patients. Blood 2020, 136, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Langerbeins, P.; Hallek, M. COVID-19 in patients with hematologic malignancy. Blood 2022, 140, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Pinana, J.L.; Martino, R.; Garcia-Garcia, I.; Parody, R.; Morales, M.D.; Benzo, G.; Gomez-Catalan, I.; Coll, R.; De La Fuente, I.; Luna, A.; et al. Risk factors and outcome of COVID-19 in patients with hematological malignancies. Exp. Hematol. Oncol. 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Salmanton-Garcia, J.; Marchesi, F.; Busca, A.; Corradini, P.; Hoenigl, M.; Klimko, N.; Koehler, P.; Pagliuca, A.; Passamonti, F.; et al. COVID-19 infection in adult patients with hematological malignancies: A European Hematology Association Survey (EPICOVIDEHA). J. Hematol. Oncol. 2021, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Hus, I.; Szymczyk, A.; Manko, J.; Drozd-Sokolowska, J. COVID-19 in Adult Patients with Hematological Malignancies-Lessons Learned after Three Years of Pandemic. Biology 2023, 12, 545. [Google Scholar] [CrossRef]
- Lekkala, M.R.; Liesveld, J. Hematologic Malignancies, Classification, and Update on Modern Interventions. In Orofacial Supportive Care in Cancer: A Contemporary Oral Oncology Perspective; Nair, R., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 11–22. [Google Scholar]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Farc, O.; Cristea, V. An overview of the tumor microenvironment, from cells to complex networks (Review). Exp. Ther. Med. 2021, 21, 96. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023. [Google Scholar] [CrossRef]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef]
- Das, K.; Prasad, R.; Ansari, S.A.; Roy, A.; Mukherjee, A.; Sen, P. Matrix metalloproteinase-2: A key regulator in coagulation proteases mediated human breast cancer progression through autocrine signaling. Biomed. Pharmacother. 2018, 105, 395–406. [Google Scholar] [CrossRef]
- Baumann, Z.; Auf der Maur, P.; Bentires-Alj, M. Feed-forward loops between metastatic cancer cells and their microenvironment-the stage of escalation. EMBO Mol. Med. 2022, 14, e14283. [Google Scholar] [CrossRef]
- Kim, H.J.; Ji, Y.R.; Lee, Y.M. Cross-talk between angiogenesis and immune regulation in the tumor microenvironment. Arch. Pharm. Res. 2022, 45, 401–416. [Google Scholar] [CrossRef]
- Chakraborty, S.; Njah, K.; Hong, W. Agrin Mediates Angiogenesis in the Tumor Microenvironment. Trends Cancer 2020, 6, 81–85. [Google Scholar] [CrossRef]
- Sun, R.; Kong, X.; Qiu, X.; Huang, C.; Wong, P.P. The Emerging Roles of Pericytes in Modulating Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 676342. [Google Scholar] [CrossRef]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef]
- Noe, J.T.; Ding, C.; Geller, A.E.; Rendon, B.E.; Yan, J.; Mitchell, R.A. A Tumor-admixture Model to Interrogate Immune Cell-dependent Tumorigenesis. Bio-Protoc. 2023, 13, e4630. [Google Scholar] [CrossRef]
- Kubelkova, K.; Bostik, V.; Joshi, L.; Macela, A. Innate Immune Recognition, Integrated Stress Response, Infection, and Tumorigenesis. Biology 2023, 12, 499. [Google Scholar] [CrossRef]
- Goff, S.L.; Danforth, D.N. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin. Breast Cancer 2021, 21, e63–e73. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Czernek, L.; Duchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Radulovic, K.; Rossini, V.; Manta, C.; Holzmann, K.; Kestler, H.A.; Niess, J.H. The early activation marker CD69 regulates the expression of chemokines and CD4 T cell accumulation in intestine. PLoS ONE 2013, 8, e65413. [Google Scholar] [CrossRef]
- Shiow, L.R.; Rosen, D.B.; Brdickova, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef]
- Smallwood, D.T.; Apollonio, B.; Willimott, S.; Lezina, L.; Alharthi, A.; Ambrose, A.R.; De Rossi, G.; Ramsay, A.G.; Wagner, S.D. Extracellular vesicles released by CD40/IL-4-stimulated CLL cells confer altered functional properties to CD4+ T cells. Blood 2016, 128, 542–552. [Google Scholar] [CrossRef]
- Chen, Z.; You, L.; Wang, L.; Huang, X.; Liu, H.; Wei, J.Y.; Zhu, L.; Qian, W. Dual effect of DLBCL-derived EXOs in lymphoma to improve DC vaccine efficacy in vitro while favor tumorgenesis in vivo. J. Exp. Clin. Cancer Res. 2018, 37, 190. [Google Scholar] [CrossRef]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8(+) T Cell Responses. Immunity 2019, 50, 738–750.e7. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Bruchard, M.; Chalmin, F.; Rebe, C. Production of adenosine by ectonucleotidases: A key factor in tumor immunoescape. J. Biomed. Biotechnol. 2012, 2012, 473712. [Google Scholar] [CrossRef]
- Busch, A.; Quast, T.; Keller, S.; Kolanus, W.; Knolle, P.; Altevogt, P.; Limmer, A. Transfer of T cell surface molecules to dendritic cells upon CD4+ T cell priming involves two distinct mechanisms. J. Immunol. 2008, 181, 3965–3973. [Google Scholar] [CrossRef]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, H.; Li, W.; Deng, Y.; Munegowda, M.A.; Chibbar, R.; Qureshi, M.; Xiang, J. Dendritic cells recruit T cell exosomes via exosomal LFA-1 leading to inhibition of CD8+ CTL responses through downregulation of peptide/MHC class I and Fas ligand-mediated cytotoxicity. J. Immunol. 2010, 185, 5268–5278. [Google Scholar] [CrossRef]
- Reiners, K.S.; Topolar, D.; Henke, A.; Simhadri, V.R.; Kessler, J.; Sauer, M.; Bessler, M.; Hansen, H.P.; Tawadros, S.; Herling, M.; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013, 121, 3658–3665. [Google Scholar] [CrossRef]
- Wang, J.; Faict, S.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Schots, R.; Vanderkerken, K.; Menu, E. Extracellular vesicle cross-talk in the bone marrow microenvironment: Implications in multiple myeloma. Oncotarget 2016, 7, 38927–38945. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Garg, T.K.; Gann, J.I.; Malaviarachchi, P.A.; Stone, K.; Macleod, V.; Greenway, A.D.; Akel, N.S.; Edmondson, R.D.; Davies, F.E.; Epstein, J.; et al. Myeloma-Derived Exosomes and Soluble Factors Suppress Natural Killer Cell Function. Blood 2016, 128, 2066. [Google Scholar] [CrossRef]
- Chillemi, A.; Quarona, V.; Antonioli, L.; Ferrari, D.; Horenstein, A.L.; Malavasi, F. Roles and Modalities of Ectonucleotidases in Remodeling the Multiple Myeloma Niche. Front. Immunol. 2017, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Chillemi, A.; Castella, B.; Schiavoni, I.; Incarnato, D.; Oliva, S.; Horenstein, A.L. CD38 and Antibody Therapy: What Can Basic Science Add? Blood 2016, 128, SCI-36. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Mancek-Keber, M.; Lainscek, D.; Bencina, M.; Chen, J.G.; Romih, R.; Hunter, Z.R.; Treon, S.P.; Jerala, R. Extracellular vesicle-mediated transfer of constitutively active MyD88(L265P) engages MyD88(wt) and activates signaling. Blood 2018, 131, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Forouzandeh Moghadam, M.; Sadeghizadeh, M. Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J. Cell. Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumor progression and cancer-associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, S.; Sun, K.; Chng, W.J. The emerging roles of exosomes in leukemogeneis. Oncotarget 2016, 7, 50698–50707. [Google Scholar] [CrossRef]
- Hong, C.S.; Muller, L.; Whiteside, T.L.; Boyiadzis, M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front. Immunol. 2014, 5, 160. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef]
- Quatromoni, J.G.; Eruslanov, E. Tumor-associated macrophages: Function, phenotype, and link to prognosis in human lung cancer. Am. J. Transl. Res. 2012, 4, 376–389. [Google Scholar]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef]
- Diao, J.; Yang, X.; Song, X.; Chen, S.; He, Y.; Wang, Q.; Chen, G.; Luo, C.; Wu, X.; Zhang, Y. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med. Oncol. 2015, 32, 453. [Google Scholar] [CrossRef]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108, djv330. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Secreto, C.R.; Knox, T.R.; Ding, W.; Mukhopadhyay, D.; Kay, N.E. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: Implications for disease progression. Blood 2010, 115, 1755–1764. [Google Scholar] [CrossRef]
- Boysen, J.; Nelson, M.; Magzoub, G.; Maiti, G.P.; Sinha, S.; Goswami, M.; Vesely, S.K.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. Dynamics of microvesicle generation in B-cell chronic lymphocytic leukemia: Implication in disease progression. Leukemia 2017, 31, 350–360. [Google Scholar] [CrossRef]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef]
- Farahani, M.; Rubbi, C.; Liu, L.; Slupsky, J.R.; Kalakonda, N. CLL Exosomes Modulate the Transcriptome and Behaviour of Recipient Stromal Cells and Are Selectively Enriched in miR-202-3p. PLoS ONE 2015, 10, e0141429. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Dempsey, C.; Chadwick, A.; Harrison, S.; Liu, J.; Di, Y.; McGinn, O.J.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P.; et al. Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood 2016, 128, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X.J.; Zeng, C.; Wu, P.H.; Wang, H.X.; Chen, Z.C.; Li, Q.B. Microvesicles secreted from human multiple myeloma cells promote angiogenesis. Acta Pharmacol. Sin. 2014, 35, 230–238. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef]
- Vardaki, I.; Sanchez, C.; Fonseca, P.; Olsson, M.; Chioureas, D.; Rassidakis, G.; Ullen, A.; Zhivotovsky, B.; Bjorkholm, M.; Panaretakis, T. Caspase-3-dependent cleavage of Bcl-xL in the stroma exosomes is required for their uptake by hematological malignant cells. Blood 2016, 128, 2655–2665. [Google Scholar] [CrossRef]
- El-Saghir, J.; Nassar, F.; Tawil, N.; El-Sabban, M. ATL-derived exosomes modulate mesenchymal stem cells: Potential role in leukemia progression. Retrovirology 2016, 13, 73. [Google Scholar] [CrossRef]
- Horiguchi, H.; Kobune, M.; Kikuchi, S.; Yoshida, M.; Murata, M.; Murase, K.; Iyama, S.; Takada, K.; Sato, T.; Ono, K.; et al. Extracellular vesicle miR-7977 is involved in hematopoietic dysfunction of mesenchymal stromal cells via poly(rC) binding protein 1 reduction in myeloid neoplasms. Haematologica 2016, 101, 437–447. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Rosón, B.; Redondo, A.; Rodríguez, C.; Rodríguez, H.; Ortega, R.; Rico, A.; Preciado, S.; López Villar, O.; et al. Extracellular Vesicles Play an Important Role in Intercellular Communication Between Bone Marrow Stroma and Hematopoietic Progenitor Cells in Myeloproliferative Neoplasms. Blood 2016, 128, 1957. [Google Scholar] [CrossRef]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef]
- Zhu, X.; You, Y.; Li, Q.; Zeng, C.; Fu, F.; Guo, A.; Zhang, H.; Zou, P.; Zhong, Z.; Wang, H.; et al. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: Implications for donor cell leukemia. Leukemia 2014, 28, 1666–1675. [Google Scholar] [CrossRef]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 2016, 7, 59173–59188. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Kumar, P.; Yadava, P.K. Telomerase and its extracurricular activities. Cell. Mol. Biol. Lett. 2013, 18, 538–554. [Google Scholar] [CrossRef]
- Mancini, S.J.C.; Balabanian, K.; Corre, I.; Gavard, J.; Lazennec, G.; Le Bousse-Kerdiles, M.C.; Louache, F.; Maguer-Satta, V.; Mazure, N.M.; Mechta-Grigoriou, F.; et al. Deciphering Tumor Niches: Lessons From Solid and Hematological Malignancies. Front. Immunol. 2021, 12, 766275. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, X.; Jiang, L.; Li, Y.; Zheng, Q. Tumor endothelial cell-derived extracellular vesicles contribute to tumor microenvironment remodeling. Cell Commun. Signal. 2022, 20, 97. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Hou, D.; Huang, Q.; Zhan, W.; Chen, C.; Liu, J.; You, R.; Xie, J.; Chen, P.; et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J. Cell. Physiol. 2019, 234, 10602–10614. [Google Scholar] [CrossRef]
- Sun, G.; Gu, Q.; Zheng, J.; Cheng, H.; Cheng, T. Emerging roles of extracellular vesicles in normal and malignant hematopoiesis. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Li, B.; Hong, J.; Hong, M.; Wang, Y.; Yu, T.; Zang, S.; Wu, Q. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene 2019, 38, 5227–5238. [Google Scholar] [CrossRef]
- Zarfati, M.; Avivi, I.; Brenner, B.; Katz, T.; Aharon, A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis 2019, 22, 185–196. [Google Scholar] [CrossRef]
- Taverna, S.; Flugy, A.; Saieva, L.; Kohn, E.C.; Santoro, A.; Meraviglia, S.; De Leo, G.; Alessandro, R. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int. J. Cancer 2012, 130, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Fontana, S.; Monteleone, F.; Pucci, M.; Saieva, L.; De Caro, V.; Cardinale, V.G.; Giallombardo, M.; Vicario, E.; Rolfo, C.; et al. Curcumin modulates chronic myelogenous leukemia exosomes composition and affects angiogenic phenotype via exosomal miR-21. Oncotarget 2016, 7, 30420–30439. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Amodeo, V.; Saieva, L.; Russo, A.; Giallombardo, M.; De Leo, G.; Alessandro, R. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer 2014, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Garnier, D.; Lee, T.H.; D’Asti, E.; Montermini, L.; Meehan, B.; Rak, J. PML-RARa modulates the vascular signature of extracellular vesicles released by acute promyelocytic leukemia cells. Angiogenesis 2016, 19, 25–38. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Dolega, M.E.; Monnier, S.; Brunel, B.; Joanny, J.F.; Recho, P.; Cappello, G. Extracellular matrix in multicellular aggregates acts as a pressure sensor controlling cell proliferation and motility. Elife 2021, 10, e63258. [Google Scholar] [CrossRef]
- Patel, N.J.; Ashraf, A.; Chung, E.J. Extracellular Vesicles as Regulators of the Extracellular Matrix. Bioengineering 2023, 10, 136. [Google Scholar] [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J. Biol. Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef]
- Redzic, J.S.; Kendrick, A.A.; Bahmed, K.; Dahl, K.D.; Pearson, C.G.; Robinson, W.A.; Robinson, S.E.; Graner, M.W.; Eisenmesser, E.Z. Extracellular vesicles secreted from cancer cell lines stimulate secretion of MMP-9, IL-6, TGF-beta1 and EMMPRIN. PLoS ONE 2013, 8, e71225. [Google Scholar] [CrossRef]
- Hansen, H.P.; Engels, H.M.; Dams, M.; Paes Leme, A.F.; Pauletti, B.A.; Simhadri, V.L.; Durkop, H.; Reiners, K.S.; Barnert, S.; Engert, A.; et al. Protrusion-guided extracellular vesicles mediate CD30 trans-signalling in the microenvironment of Hodgkin’s lymphoma. J. Pathol. 2014, 232, 405–414. [Google Scholar] [CrossRef]
- Mugnaini, E.N.; Ghosh, N. Lymphoma. Prim. Care Clin. Off. Pract. 2016, 43, 661–675. [Google Scholar] [CrossRef]
- Cordone, I.; Masi, S.; Giannarelli, D.; Pasquale, A.; Conti, L.; Telera, S.; Pace, A.; Papa, E.; Marino, M.; de Fabritiis, P.; et al. Major Differences in Lymphocyte Subpopulations Between Cerebrospinal Fluid and Peripheral Blood in Non-Hodgkin Lymphoma Without Leptomeningeal Involvement: Flow Cytometry Evidence of a Cerebral Lymphatic System. Front. Oncol. 2021, 11, 685786. [Google Scholar] [CrossRef]
- Kulka, M.; Brennan, K.; Mc Gee, M. Investigation of canine extracellular vesicles in diffuse large B-cell lymphomas. PLoS ONE 2022, 17, e0274261. [Google Scholar] [CrossRef]
- Matthiesen, R.; Gameiro, P.; Henriques, A.; Bodo, C.; Moraes, M.C.S.; Costa-Silva, B.; Cabecadas, J.; Gomes da Silva, M.; Beck, H.C.; Carvalho, A.S. Extracellular Vesicles in Diffuse Large B Cell Lymphoma: Characterization and Diagnostic Potential. Int. J. Mol. Sci. 2022, 23, 3327. [Google Scholar] [CrossRef]
- Higuchi, H.; Yamakawa, N.; Imadome, K.I.; Yahata, T.; Kotaki, R.; Ogata, J.; Kakizaki, M.; Fujita, K.; Lu, J.; Yokoyama, K.; et al. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood 2018, 131, 2552–2567. [Google Scholar] [CrossRef]
- Yang, J.; Li, W.; He, X.; Zhang, G.; Yue, L.; Chai, Y. VEGF overexpression is a valuable prognostic factor for non-Hodgkin’s lymphoma evidence from a systemic meta-analysis. Dis. Markers 2015, 2015, 786790. [Google Scholar] [CrossRef]
- Kudo, K.; Miki, Y.; Carreras, J.; Nakayama, S.; Nakamoto, Y.; Ito, M.; Nagashima, E.; Yamamoto, K.; Higuchi, H.; Morita, S.Y.; et al. Secreted phospholipase A(2) modifies extracellular vesicles and accelerates B cell lymphoma. Cell Metab. 2022, 34, 615–633.e618. [Google Scholar] [CrossRef]
- Dorsam, B.; Bosl, T.; Reiners, K.S.; Barnert, S.; Schubert, R.; Shatnyeva, O.; Zigrino, P.; Engert, A.; Hansen, H.P.; von Strandmann, E.P. Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front. Immunol. 2018, 9, 1358. [Google Scholar] [CrossRef]
- Maltoni, F.; Ciardiello, C.; Nanni, L.; Barone, M.; Narimanfar, G.; Casadei, B.; Argnani, L.; Broccoli, A.; Forte, D.; Budillon, A.; et al. Critical Role of Hodgkin Lymphoma Cells-Derived Large and Small Extracellular Vesicles in the Regulation of the Immune-Inflammatory Microenvironment. Blood 2022, 140, 11509–11511. [Google Scholar] [CrossRef]
- Repetto, O.; Lovisa, F.; Elia, C.; Enderle, D.; Romanato, F.; Buffardi, S.; Sala, A.; Pillon, M.; Steffan, A.; Burnelli, R.; et al. Proteomic Exploration of Plasma Exosomes and Other Small Extracellular Vesicles in Pediatric Hodgkin Lymphoma: A Potential Source of Biomarkers for Relapse Occurrence. Diagnostics 2021, 11, 917. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.C.; Fachel, A.A.; Li, S.; Sawh, S.; Muley, A.; Ishii, J.; Saxena, A.; Dominguez, P.M.; Caldas Lopes, E.; Agirre, X.; et al. Extracellular vesicles in DLBCL provide abundant clues to aberrant transcriptional programming and genomic alterations. Blood 2018, 132, e13–e23. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Shi, D.; Wan, X.C.; Hu, J.; Su, Y.F.; Zeng, Y.P.; Hu, Z.J.; Yu, B.H.; Zhang, Q.L.; Wei, P.; et al. Universal extracellular vesicles and PD-L1+ extracellular vesicles detected by single molecule array technology as circulating biomarkers for diffuse large B cell lymphoma. Oncoimmunology 2021, 10, 1995166. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, S.; Zhou, T.; Li, X.; Tang, J. Potential of the tumor-derived extracellular vesicles carrying the miR-125b-5p target TNFAIP3 in reducing the sensitivity of diffuse large B cell lymphoma to rituximab. Int. J. Oncol. 2021, 58, 31. [Google Scholar] [CrossRef]
- Tang, J.; Hu, P.; Zhou, S.; Zhou, T.; Li, X.; Zhang, L. Lymphoma cell-derived extracellular vesicles inhibit autophagy and apoptosis to promote lymphoma cell growth via the microRNA-106a/Beclin1 axis. Cell Cycle 2022, 21, 1280–1293. [Google Scholar] [CrossRef]
- Tickner, J.A.; Urquhart, A.J.; Stephenson, S.A.; Richard, D.J.; O’Byrne, K.J. Functions and therapeutic roles of exosomes in cancer. Front. Oncol. 2014, 4, 127. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- van Doormaal, F.F.; Kleinjan, A.; Di Nisio, M.; Buller, H.R.; Nieuwland, R. Cell-derived microvesicles and cancer. Neth. J. Med. 2009, 67, 266–273. [Google Scholar]
- Zocco, D.; Ferruzzi, P.; Cappello, F.; Kuo, W.P.; Fais, S. Extracellular vesicles as shuttles of tumor biomarkers and anti-tumor drugs. Front. Oncol. 2014, 4, 267. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Filipazzi, P.; Burdek, M.; Villa, A.; Rivoltini, L.; Huber, V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin. Cancer Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef]
- Caivano, A.; Laurenzana, I.; De Luca, L.; La Rocca, F.; Simeon, V.; Trino, S.; D’Auria, F.; Traficante, A.; Maietti, M.; Izzo, T.; et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol. 2015, 36, 9739–9752. [Google Scholar] [CrossRef]
- Hong, C.S.; Muller, L.; Boyiadzis, M.; Whiteside, T.L. Isolation and characterization of CD34+ blast-derived exosomes in acute myeloid leukemia. PLoS ONE 2014, 9, e103310. [Google Scholar] [CrossRef]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef]
- Caivano, A.; La Rocca, F.; Simeon, V.; Girasole, M.; Dinarelli, S.; Laurenzana, I.; De Stradis, A.; De Luca, L.; Trino, S.; Traficante, A.; et al. MicroRNA-155 in serum-derived extracellular vesicles as a potential biomarker for hematologic malignancies—A short report. Cell. Oncol. 2017, 40, 97–103. [Google Scholar] [CrossRef]
- Ohyashiki, K.; Umezu, T.; Katagiri, S.; Kobayashi, C.; Azuma, K.; Tauchi, T.; Okabe, S.; Fukuoka, Y.; Ohyashiki, J.H. Downregulation of Plasma miR-215 in Chronic Myeloid Leukemia Patients with Successful Discontinuation of Imatinib. Int. J. Mol. Sci. 2016, 17, 570. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Luk, F.; Brown, R.D.; Suen, H.; Kwan, Y.; Bebawy, M. Isolation of Human CD138(+) Microparticles from the Plasma of Patients with Multiple Myeloma. Neoplasia 2016, 18, 25–32. [Google Scholar] [CrossRef]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Agarwal, K.; Branson, O.E.; Rocci, A.; Cordero, H.; Phelps, M.A.; Hade, E.M.; Dubovsky, J.A.; et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 2016, 136, 89–98. [Google Scholar] [CrossRef]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; D’Arena, G.; Simeon, V.; Trino, S.; Laurenzana, I.; Caivano, A.; La Rocca, F.; Villani, O.; Mansueto, G.; Deaglio, S.; et al. Characterization and prognostic relevance of circulating microvesicles in chronic lymphocytic leukemia. Leuk. Lymphoma 2017, 58, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Belov, L.; Matic, K.J.; Hallal, S.; Best, O.G.; Mulligan, S.P.; Christopherson, R.I. Extensive surface protein profiles of extracellular vesicles from cancer cells may provide diagnostic signatures from blood samples. J. Extracell. Vesicles 2016, 5, 25355. [Google Scholar] [CrossRef] [PubMed]
- Ferrajoli, A.; Shanafelt, T.D.; Ivan, C.; Shimizu, M.; Rabe, K.G.; Nouraee, N.; Ikuo, M.; Ghosh, A.K.; Lerner, S.; Rassenti, L.Z.; et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood 2013, 122, 1891–1899. [Google Scholar] [CrossRef]
- van Eijndhoven, M.A.; Zijlstra, J.M.; Groenewegen, N.J.; Drees, E.E.; van Niele, S.; Baglio, S.R.; Koppers-Lalic, D.; van der Voorn, H.; Libregts, S.F.; Wauben, M.H.; et al. Plasma vesicle miRNAs for therapy response monitoring in Hodgkin lymphoma patients. JCI Insight 2016, 1, e89631. [Google Scholar] [CrossRef]
- Bouyssou, J.M.C.; Aljawai, Y.; Yosef, A.; Manier, S.; Rawal, R.; Kokubun, K.; Perilla Glen, A.; Sacco, A.; Castillo, J.J.; Treon, S.P.; et al. Profiling of Circulating Exosomes in Patients with Waldenström Macroglobulinemia. Blood 2016, 128, 2940. [Google Scholar] [CrossRef]
- Cesi, G.; Walbrecq, G.; Margue, C.; Kreis, S. Transferring intercellular signals and traits between cancer cells: Extracellular vesicles as “homing pigeons”. Cell Commun. Signal. 2016, 14, 13. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef]
- Efferth, T.; Gillet, J.P.; Sauerbrey, A.; Zintl, F.; Bertholet, V.; de Longueville, F.; Remacle, J.; Steinbach, D. Expression profiling of ATP-binding cassette transporters in childhood T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2006, 5, 1986–1994. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef]
- Lee, C.H. Reversing agents for ATP-binding cassette drug transporters. Methods Mol. Biol. 2010, 596, 325–340. [Google Scholar] [CrossRef]
- Koch, R.; Aung, T.; Vogel, D.; Chapuy, B.; Wenzel, D.; Becker, S.; Sinzig, U.; Venkataramani, V.; von Mach, T.; Jacob, R.; et al. Nuclear Trapping through Inhibition of Exosomal Export by Indomethacin Increases Cytostatic Efficacy of Doxorubicin and Pixantrone. Clin. Cancer Res. 2016, 22, 395–404. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.; van Meerloo, J.; Assaraf, Y.G.; et al. Exosomes Secreted by Apoptosis-Resistant Acute Myeloid Leukemia (AML) Blasts Harbor Regulatory Network Proteins Potentially Involved in Antagonism of Apoptosis. Mol. Cell. Proteom. 2016, 15, 1281–1298. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogne, J.M. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schon, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B.; et al. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with Brentuximab-Vedotin, in vitro. Oncotarget 2016, 7, 30523–30535. [Google Scholar] [CrossRef]
- McKiernan, J.; Donovan, M.J.; Margolis, E.; Partin, A.; Carter, B.; Brown, G.; Torkler, P.; Noerholm, M.; Skog, J.; Shore, N.; et al. A Prospective Adaptive Utility Trial to Validate Performance of a Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer in Patients with Prostate-specific Antigen 2–10ng/ml at Initial Biopsy. Eur. Urol. 2018, 74, 731–738. [Google Scholar] [CrossRef]
- Wang, W.W.; Sorokin, I.; Aleksic, I.; Fisher, H.; Kaufman, R.P., Jr.; Winer, A.; McNeill, B.; Gupta, R.; Tilki, D.; Fleshner, N.; et al. Expression of Small Noncoding RNAs in Urinary Exosomes Classifies Prostate Cancer into Indolent and Aggressive Disease. J. Urol. 2020, 204, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Garrastacho, M.; Bajo-Santos, C.; Line, A.; Martens-Uzunova, E.S.; de la Fuente, J.M.; Moros, M.; Soekmadji, C.; Tasken, K.A.; Llorente, A. Extracellular vesicles as a source of prostate cancer biomarkers in liquid biopsies: A decade of research. Br. J. Cancer 2022, 126, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.M.; Zhang, C.; Wang, Z.; Ni, Z.; Zhang, S.; Yang, S.; Huang, X.; Mo, L.; Li, J.; Lee, B.; et al. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 2021, 3, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Shaba, E.; Vantaggiato, L.; Governini, L.; Haxhiu, A.; Sebastiani, G.; Fignani, D.; Grieco, G.E.; Bergantini, L.; Bini, L.; Landi, C. Multi-Omics Integrative Approach of Extracellular Vesicles: A Future Challenging Milestone. Proteomes 2022, 10, 12. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Urabe, F.; Kosaka, N.; Ito, K.; Kimura, T.; Egawa, S.; Ochiya, T. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am. J. Physiol. Cell Physiol. 2020, 318, C29–C39. [Google Scholar] [CrossRef]
- Wang, Z.; Mo, H.; He, Z.; Chen, A.; Cheng, P. Extracellular vesicles as an emerging drug delivery system for cancer treatment: Current strategies and recent advances. Biomed. Pharmacother. 2022, 153, 113480. [Google Scholar] [CrossRef]





| Type | Sub-Class | Sub-Sub-Class | Characteristics |
|---|---|---|---|
| Myeloid neoplasm | MPN | CML | Excessive granulocyte proliferation leading to increased myeloid maturation |
| Classic BCR:ABL1-negative MPN: (1) PV | Increased red blood cells | ||
| (2) ET | Clonal disorder related to thrombocytosis | ||
| (3) PF | Polyclonal increase in fibroblasts leading to BM fibrosis | ||
| CNL | Overproduction of matured granulocytes | ||
| CEL | Clonal proliferation of abnormal eosinophils resulting in hypereosinophilia | ||
| Myeloproliferative neoplasm, unclassifiable | Does not have specific MPN characteristics or possess overlapping MPN features | ||
| MDS | According to genetic abnormalities: (1) Low blasts and isolated del(5q) | 5q deletion, BMB < 5% in BM, and PB < 2%, SF3B1/TP53 mutation | |
| (2) Low blasts and SF3B1 mutation | BMB < 5% and PB < 2%, SF3B1 mutation, ≥15% ring sideroblasts | ||
| (3) Biallelic TP53 inactivation | BMB and PB < 20%; copy number loss/neutral results in loss of heterozygosity, complex cytogenetics | ||
| According to morphology: (1) Low blasts | BMB < 5% and PB < 2% | ||
| (2) Hypoblastic | BM cellularity < 20% | ||
| (3) MDS-IB | MDS-IB1: BMB 5–9% and PB 2–4%; MDS-IB2; BMB 10–19% and PB 5–19%; MDS with increased blasts and fibrosis: BM fibrosis with BMB 5–19% and PB 2–19% | ||
| AML | AML with recurring genetic abnormalities | Categorized into characteristic chromosomal rearrangements and no karyotypic abnormalities | |
| AML with myelodysplasia-related features | Lacks genetic features of AML but with history of MDS or MDS/MPN | ||
| Therapy-related AML | Arises after chemo- or radiation therapy | ||
| AML-NOS | With clinical, morphologic, and immunophenotypic features but lacking AML diagnostic karyotype | ||
| Myeloid sarcoma | Tumor mass outside BM effaces into tissues | ||
| Myeloid proliferation related to DS | DS, associated with increased AML | ||
| Myeloid neoplasm with mutated TP53 | MDS-mutated TP53 | <10% blasts | |
| MDS/AML-mutated TP53 | 10–19% blasts | ||
| AML-mutated TP53 | >20% blasts | ||
| Lymphoid neoplasm | Precursor lymphoid neoplasm | B-cell ALL/LBL | Arises from B-cell precursor, representing most ALL/LBL |
| T-cell ALL/LBL | Arises from T-cell precursor, representing 15% ALL/LBL | ||
| Mature B-cell neoplasm | CLL/SLL | Small, mature-appearing lymphocytes | |
| LPL | Derived from post-germinal center of B-cells | ||
| Monoclonal gammopathy | A type of lymphoid neoplasm for primary cold agglutinin disease | ||
| Plasma cell neoplasm | Associated with terminally differentiated germinal centers of B-cells | ||
| Hairy cell leukemia | Associated with post-germinal centers of B-cells | ||
| MZL | Associated with marginal zone of matured B-cells | ||
| FL: (1) Classic FL | Proliferation of centrocytes and centroblasts | ||
| (2) FL grade 3B | Uncontrolled proliferation of centroblasts | ||
| (3) FL-unusual feature | Comprising FL with blastoid features and FL with diffused growth pattern | ||
| MCL | Neoplasm of naïve and antigen-presenting B-cells | ||
| DLBCL | Neoplasm of matured B-cells | ||
| High-grade B-cell lymphoma | Comprising aggressive BL and other high-grade B-cell lymphoma | ||
| HL | cHL | Germinal and post-germinal centers of B-cells; again classified as nodular sclerosis cHL, mixed cellularity cHL, and lymphocyte rich- or lymphocyte-depleted cHL | |
| NLPHL | Associated with germinal neoplastic B-cells | ||
| Matured T-cell/NK-cell lineage lymphoma | PTCL: (1) PTCL-NOS | Not fitting the criteria of T-cell lymphomas | |
| (2) ALCL | According to the expression of ALK, sub-categorized into “ALCL, ALK-positive” or “ALCL, ALK-negative” and “breast implant-associated ALCL.” | ||
| (3) Follicular TH | Comprising AITL and related follicular TH cell lymphomas | ||
| (4) Extranodal | EBV-associated neoplasm arises in lymph nodes | ||
| NK/T-cell lymphoma nasal type | |||
| (5) Hepatosplenic T-cell lymphoma | Aggressive and related to immunosuppression | ||
| (6) Primary intestinal T-cell lymphoma | Aggressive T-cell lymphoma in intestinal tract | ||
| Primary cutaneous T-cell lymphoma | Lymphoma of cutaneous peripheral T-cells | ||
| ATL | Associated with peripheral T-cells from HTLV-1 infected CD4+ T-cells | ||
| TLGL leukemia | Derived from clonally expanded granular T-cells | ||
| T-cell polymorphocytic leukemia | Highly aggressive, medium-sized matured T-cells | ||
| NKLGL leukemia | Aggressive, associated with malignant NK-cells | ||
| Aggressive | Aggressive, associated with malignant NK-cells | ||
| Aggressive | Nasal type, associated with EBV infection | ||
| NK-cell leukemia | |||
| Myeloid and lymphoid neoplasm with eosinophilia and TK gene fusion | HES | Primary (or neoplastic) | Clonal eosinophilic expansion reaching underlying stem cells and myeloid and eosinophilic neoplasm |
| Secondary (or reactive) | Polyclonal eosinophilic expansion mediated by overproduction of eosinophilic cytokines during parasitic infections; certain solid tumor T-cell lymphomas | ||
| Idiopathic | The underlying cause of HE remains unknown | ||
| Specific syndrome associated with HE | - | Disease complications and clinical presentation are not fully defined, e.g., EGPA and some immunodeficiencies | |
| HEUS | - | Persistent unexplained HE; difficult to predict whether the patients will develop clinical manifestations leading to HES | |
| Mastocytosis | SM | Indolent SM | 70% of SM is found to be indolent SM, which may or may not cause skin lesions of maculopapular cutaneous mastocytosis (MPCM) |
| Smoldering SM | Rare, shows same phenotypic effects as indolent SM | ||
| Aggressive SM | Highly aggressive, manifesting round, rather than spindle-shaped, mast cells with median survival from month to years | ||
| MCL | Same phenotypic responses as aggressive SM | ||
| SM-AHN | Similar to SM phenotype but requiring urgent treatment depending on the disease stage in BM and extracutaneous sites | ||
| Histiocytic or dendritic neoplasm | Histiocytic sarcoma | - | Malignant proliferation of cells with morphologic and immunophenotypic features of mature tissue histiocytes |
| Tumors of Langerhans cells | Langerhans cell histiocytosis | Malignancies of cells expressing CD1a, langerin, and S100 | |
| Langerhans cell sarcoma | High-grade malignancy with same features as Langerhans cell histiocytosis | ||
| Indeterminate dendritic cell tumor | - | Rare, involving proliferation of cells, spindle-shaped to ovoid, similar to IDC | |
| Interdigitating dendritic cell sarcoma | Very rare, involving spindle to ovoid cells resembling interdigitating dendritic cells | ||
| Follicular dendritic cell sarcoma | Neoplastic proliferation of spindle to ovoid cells; morphologically and immunophenotypically similar to follicular dendritic cells | ||
| Inflammatory pseudotumor-like follicular/fibroblastic dendritic cell sarcoma | Neoplastic spindle cells residing in lymphoplasmacytic infiltrate involving liver and spleen | ||
| Fibroblastic reticular cell tumor | Very rare, spindle cells with cytokeratin involving skin, spleen, and lymph nodes | ||
| Disseminated juvenile xanthogranuloma | Proliferation and dissemination of small oval histiocytes resembling dermal juvenile xanthogranuloma of skin and soft tissues | ||
| Erdheim–Chester disease | Involving foamy histiocytes of bones leading to clonal proliferation and associated with heart, CNS, and retroperitoneum | ||
| Mixed myeloid and lymphoid neoplasm | MPAL | Ph+ MPAL | With Philadelphia chromosome (Ph) |
| Ph− categories: (1) MPAL with t(v;11q23.3); KMT2A-rearranged | MPAL associated with detectable t(v;11q23.3) and KMT2A rearrangement | ||
| (2) MPAL B/myeloid NOS | MPAL with B-lymphoblast immunophenotype lacking Ph chromosome or t(v;11q23.3) | ||
| (3) MPAL T/myeloid, NOS | MPAL with T-lymphoblast immunophenotype lacking Ph chromosome or t(v;11q23.3) |
| Disease | EVs’ Biomarker | Up-/Down-Regulation | References |
|---|---|---|---|
| AML | TGF-β | Up | [189] |
| CD13, CD117, CD34 | Up | [195,196] | |
| FLT-ITD, NPM1, IGF-IR, CXCR4, MMP4 mRNA | - | [197] | |
| miR-155 | Up | [198] | |
| CML | CD13 | Up | [195] |
| miR-215 | Up | [199] | |
| MM | CD38, CD138, CD147 | Up | [66,195,200] |
| CD44 | Up | [201] | |
| let-7b, miR-18a | Down | [202] | |
| CLL | CD19, CD37 | Up | [203] |
| CD52 | Up | [139] | |
| CD19, CD5, CD44, CD31, CD55, CD82, CD62L, HLA-A, B, C, HLA-DR | Up | [204] | |
| CD49c, CD21, CD63 | Down | [204] | |
| miR-155 | Up | [205] | |
| HL | CD30 | Down | [195] |
| miR-155, miR-127, miR-21, let-7 | Up | [195,206] | |
| WL | miR-155 | Up | [195,207] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, K.; Mukherjee, T.; Shankar, P. The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy. Biomolecules 2023, 13, 897. https://doi.org/10.3390/biom13060897
Das K, Mukherjee T, Shankar P. The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy. Biomolecules. 2023; 13(6):897. https://doi.org/10.3390/biom13060897
Chicago/Turabian StyleDas, Kaushik, Tanmoy Mukherjee, and Prem Shankar. 2023. "The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy" Biomolecules 13, no. 6: 897. https://doi.org/10.3390/biom13060897
APA StyleDas, K., Mukherjee, T., & Shankar, P. (2023). The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy. Biomolecules, 13(6), 897. https://doi.org/10.3390/biom13060897