

Aldosterone: Essential for Life but Damaging to the Vascular Endothelium


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
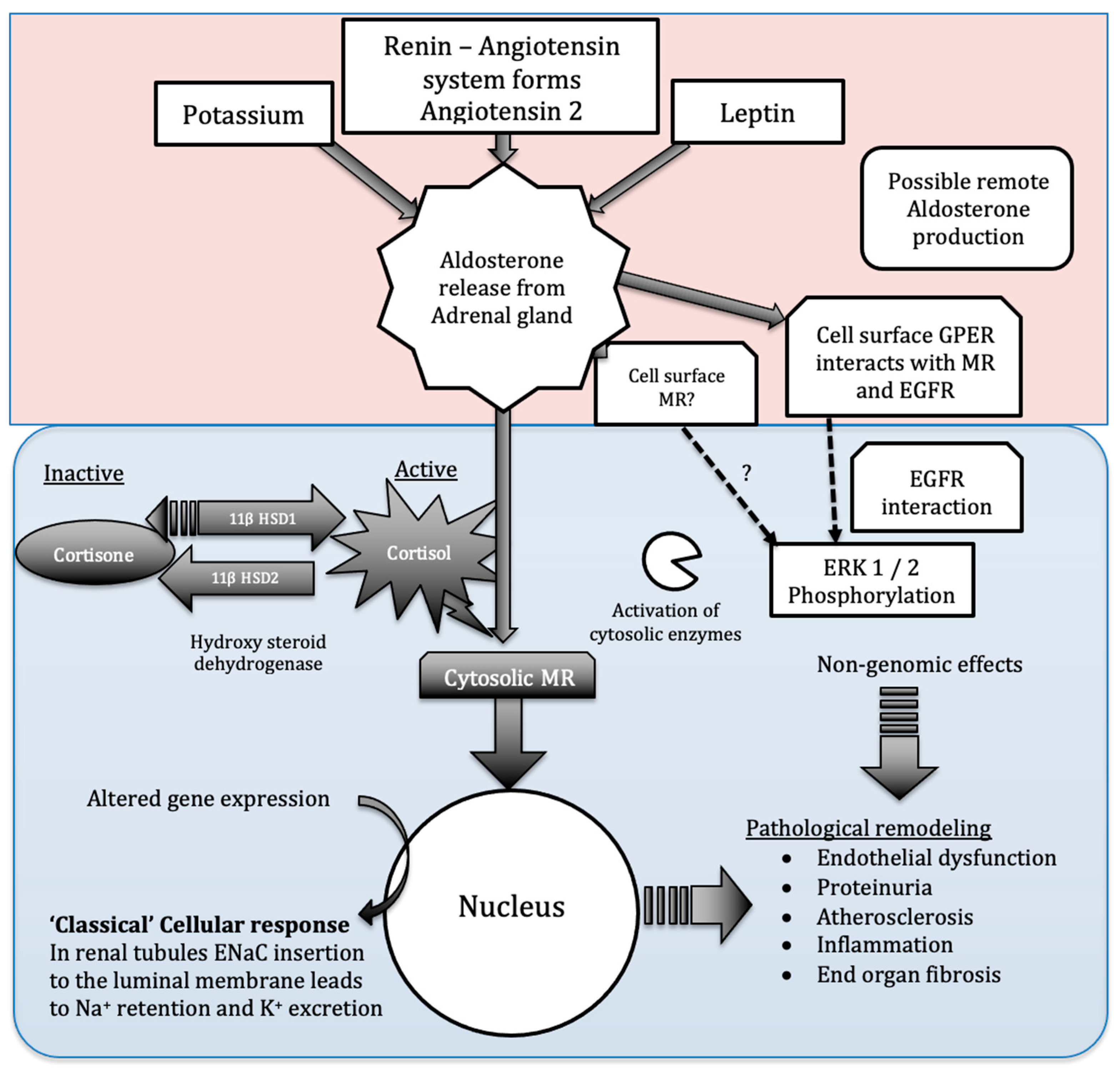
2. Aldosterone Production
3. Aldosterone Acts on Multiple Tissues and May Act via Multiple Receptors
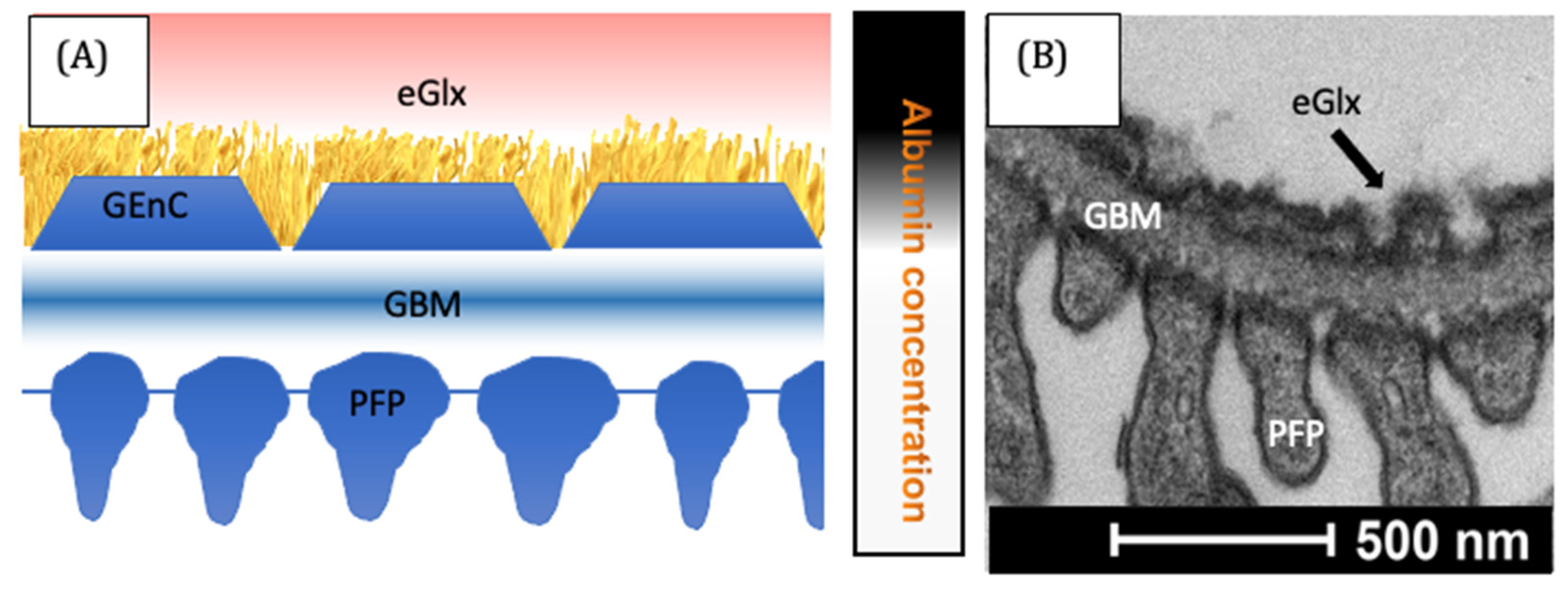
4. The Vascular Endothelium
5. Aldosterone Disrupts Flow-Mediated Dilatation
6. Aldosterone Increases Permeability to Macromolecules
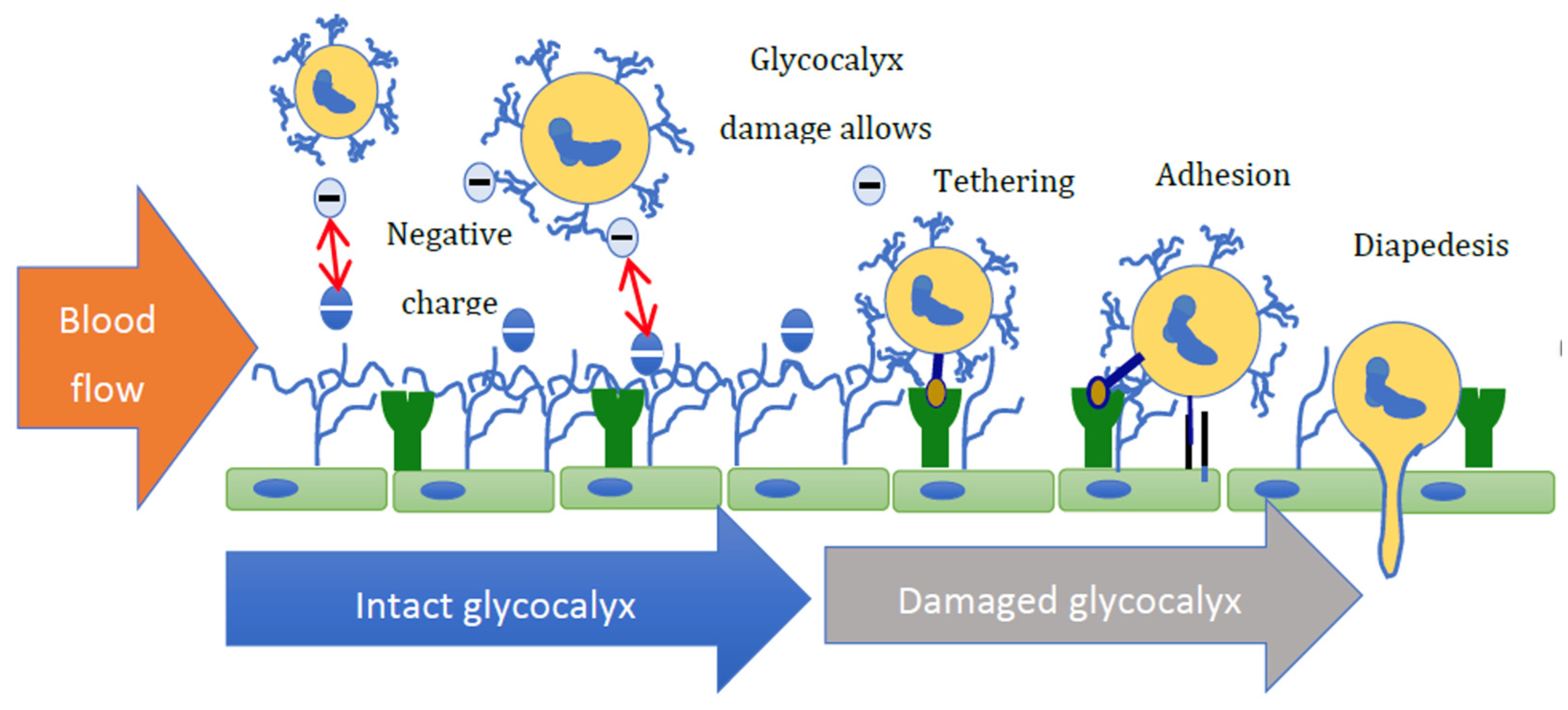
7. Aldosterone Promotes Inflammation
8. Aldosterone Promotes Atherosclerosis
9. Pharmacological Manipulation of the Aldosterone—MR System
9.1. Angiotensin Inhibition
9.2. Mineralocorticoid Antagonism
9.3. Aldosterone Synthase Inhibitors
10. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 11 β-HSD1 | 11 –beta-hydroxysteroid dehydrogenase type 1 |
| 11 β-HSD2 | 11 –beta-hydroxysteroid dehydrogenase type 2 |
| ACE | angiotensin converting enzyme |
| ACEi | angiotensin-converting enzyme inhibitor |
| ADPKD | autosomal dominant polycystic kidney disease |
| AKI | acute kidney injury |
| AMPK | AMP-activated protein kinase |
| ApoE | Apolipoprotein E |
| AR | androgen receptor |
| ARB | angiotensin receptor blockers |
| BP | blood pressure |
| CHF | congestive heart failure |
| CKD | chronic kidney disease |
| CNS | central nervous system |
| CYP11B1 | cytochrome P450 family 11 subfamily B member 1 |
| CYP11B2 | cytochrome P450 family 11 subfamily B member 2 |
| DOCA | deoxycorticosterone acetate |
| EGFR | epidermal growth factor receptor |
| ENaC | epithelial sodium channels |
| EnC | endothelial cell |
| eGlx | endothelial cell glycocalyx |
| eNOS | endothelial nitric oxide synthase |
| ERK 1/2 | extracellular signal-regulated kinase 1/2 |
| ET-1 | endothelin-1 |
| FMD | flow-mediated dilatation |
| GPER (or GPR 30) | G-protein associated oestrogen receptor |
| HAC | human adrenocortical carcinoma |
| HEK | human embryonic kidney |
| HS | heparan sulphate |
| HUVEC | human umbilical vein endothelial cells |
| HYAL2 | hyaluronidase 2 |
| ICAM-1 | intercellular adhesion molecule-1 |
| MMP | matrix metalloproteinase |
| MR | mineralocorticoid receptor |
| NHE | Na+-H+ exchanger |
| NO | nitric oxide |
| ox-LDL | oxidized low-density lipoprotein |
| PR | progesterone receptor |
| RAAS | renin angiotensin aldosterone system |
| RCT | randomized controlled trial |
| SGK-1 | serum and glucocorticoid- induced kinase 1 |
| siRNA | small interfering RNA |
| St AR Protein | steroidogenic acute regulatory protein |
| VCAM-1 | vascular cell adhesion molecule-1 |
References
- Rossier, B.C.; Baker, M.E.; Studer, R.A. Epithelial sodium transport and its control by aldosterone: The story of our internal environment revisited. Physiol. Rev. 2015, 95, 297–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kem, D.C.; Weinberger, M.H.; Gomez-Sanchez, C.; Kramer, N.J.; Lerman, R.; Furuyama, S.; Nugent, C.A. Circadian rhythm of plasma aldosterone concentration in patients with primary aldosteronism. J. Clin. Investig. 1973, 52, 2272–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, C.; Holland, O.B.; Higgins, J.R.; Kem, D.C.; Kaplan, N.M. Circadian rhythms of serum renin activity and serum corticosterone, prolactin, and aldosterone concentrations in the male rat on normal and low-sodium diets. Endocrinology 1976, 99, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Colussi, G.; Nadalini, E.; Chiuch, A.; Baroselli, S.; Lapenna, R.; Sechi, L.A. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch. Intern. Med. 2008, 168, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yin, G.S.; Tang, J.Y.; Ma, D.J.; Ru, J.; Huang, X.H. Endothelial dysfunction in patients with primary aldosteronism: A biomarker of target organ damage. J. Hum. Hypertens. 2014, 28, 711–715. [Google Scholar] [CrossRef]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F.L. Pathogenesis and treatment of primary aldosteronism. Nat. Rev. Endocrinol. 2020, 16, 578–589. [Google Scholar] [CrossRef]
- Shibata, H.; Itoh, H. Mineralocorticoid receptor-associated hypertension and its organ damage: Clinical relevance for resistant hypertension. Am. J. Hypertens. 2012, 25, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Bomback, A.S.; Klemmer, P.J. Renal injury in extreme obesity: The important role of aldosterone. Kidney Int. 2008, 74, 1216, author reply 1216–1217. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M. Activation of the aldosterone/mineralocorticoid receptor system in chronic kidney disease and metabolic syndrome. Clin. Exp. Nephrol. 2010, 14, 303–314. [Google Scholar] [CrossRef]
- Bomback, A.S.; Kshirsagar, A.V.; Ferris, M.E.; Klemmer, P.J. Disordered aldosterone-volume relationship in end-stage kidney disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2009, 10, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Adeseun, G.A.; Rosas, S.E. The impact of obstructive sleep apnea on chronic kidney disease. Curr. Hypertens. Rep. 2010, 12, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Nagasawa, Y.; Iwatani, H.; Shinzawa, M.; Obi, Y.; Teranishi, J.; Ishigami, T.; Yamauchi-Takihara, K.; Nishida, M.; Rakugi, H.; et al. Self-reported sleep duration and prediction of proteinuria: A retrospective cohort study. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2012, 59, 343–355. [Google Scholar] [CrossRef]
- Hattangady, N.G.; Olala, L.O.; Bollag, W.B.; Rainey, W.E. Acute and chronic regulation of aldosterone production. Mol. Cell. Endocrinol. 2012, 350, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemele, E.J. The Adipocyte-Derived Hormone Leptin is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef]
- Fujisaki, M.; Nagoshi, T.; Nishikawa, T.; Date, T.; Yoshimura, M. Rapid induction of aldosterone synthesis in cultured neonatal rat cardiomyocytes under high glucose conditions. BioMed Res. Int. 2013, 2013, 161396. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, S.M.; Connell, J.M.; Davies, E. Non-adrenal synthesis of aldosterone: A reality check. Mol. Cell. Endocrinol. 2012, 350, 163–167. [Google Scholar] [CrossRef]
- Maron, B.A.; Oldham, W.M.; Chan, S.Y.; Vargas, S.O.; Arons, E.; Zhang, Y.Y.; Loscalzo, J.; Leopold, J.A. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 2014, 130, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Miyamori, I.; Yoneda, T.; Hatakeyama, H.; Inaba, S.; Furukawa, K.; Mabuchi, H.; Takeda, R. Regulation of aldosterone synthase in human vascular endothelial cells by angiotensin II and adrenocorticotropin. J. Clin. Endocrinol. Metab. 1996, 81, 2797–2800. [Google Scholar] [CrossRef]
- Ahmad, N.; Romero, D.G.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Do human vascular endothelial cells produce aldosterone? Endocrinology 2004, 145, 3626–3629. [Google Scholar] [CrossRef] [Green Version]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Correa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, D.M.; Shaqura, M.; Li, X.; Shakibaei, M.; Beyer, A.; Treskatsch, S.; Schafer, M.; Mousa, S.A. Aldosterone Synthase in Peripheral Sensory Neurons Contributes to Mechanical Hypersensitivity during Local Inflammation in Rats. Anesthesiology 2020, 132, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.T. Aldosteronism revisited: Perspectives on less well-recognized actions of aldosterone. J. Lab. Clin. Med. 2003, 142, 71–82. [Google Scholar] [CrossRef] [PubMed]
- McGraw, A.P.; McCurley, A.; Preston, I.R.; Jaffe, I.Z. Mineralocorticoid receptors in vascular disease: Connecting molecular pathways to clinical implications. Curr. Atheroscler. Rep. 2013, 15, 340. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, G.; Pizard, A.; Gueret, A.; El Moghrabi, S.; Ouvrard-Pascaud, A.; Berger, S.; Challande, P.; Jaffe, I.Z.; Labat, C.; Lacolley, P.; et al. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 2014, 63, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Koenig, J.B.; Jaffe, I.Z. Direct role for smooth muscle cell mineralocorticoid receptors in vascular remodeling: Novel mechanisms and clinical implications. Curr. Hypertens. Rep. 2014, 16, 427. [Google Scholar] [CrossRef] [Green Version]
- Tarjus, A.; Belozertseva, E.; Louis, H.; El Moghrabi, S.; Labat, C.; Lacolley, P.; Jaisser, F.; Galmiche, G. Role of smooth muscle cell mineralocorticoid receptor in vascular tone. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1643–1650. [Google Scholar] [CrossRef]
- Chadwick, J.A.; Hauck, J.S.; Lowe, J.; Shaw, J.J.; Guttridge, D.C.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Rafael-Fortney, J.A. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4544–4554. [Google Scholar] [CrossRef] [Green Version]
- Gilet, A.; Zou, F.; Boumenir, M.; Frippiat, J.P.; Thornton, S.N.; Lacolley, P.; Ropars, A. Aldosterone up-regulates MMP-9 and MMP-9/NGAL expression in human neutrophils through p38, ERK1/2 and PI3K pathways. Exp. Cell. Res. 2015, 331, 152–163. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Dinh Cat, A.; Griol-Charhbili, V.; Loufrani, L.; Labat, C.; Benjamin, L.; Farman, N.; Lacolley, P.; Henrion, D.; Jaisser, F. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2454–2463. [Google Scholar] [CrossRef] [Green Version]
- Lang, F. Stiff endothelial cell syndrome in vascular inflammation and mineralocorticoid excess. Hypertension 2011, 57, 146–147. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Sekizawa, N.; Yoshimoto, T.; Hayakawa, E.; Suzuki, N.; Sugiyama, T.; Hirata, Y. Transcriptome analysis of aldosterone-regulated genes in human vascular endothelial cell lines stably expressing mineralocorticoid receptor. Mol. Cell. Endocrinol. 2011, 341, 78–88. [Google Scholar] [CrossRef]
- Bienvenu, L.A.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Mineralocorticoid receptors and the heart, multiple cell types and multiple mechanisms: A focus on the cardiomyocyte. Clin. Sci. 2013, 125, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Lother, A.; Moser, M.; Bode, C.; Feldman, R.D.; Hein, L. Mineralocorticoids in the heart and vasculature: New insights for old hormones. Annu. Rev. Pharm. Toxicol. 2015, 55, 289–312. [Google Scholar] [CrossRef]
- Armani, A.; Marzolla, V.; Fabbri, A.; Caprio, M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J. Mol. Endocrinol. 2015, 55, R1–R10. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.J.; Ramnath, R.; Kadoya, H.; Desposito, D.; Riquier-Brison, A.; Ferguson, J.K.; Onions, K.L.; Ogier, A.S.; ElHegni, H.; Coward, R.J.; et al. Aldosterone induces albuminuria via matrix metalloproteinase-dependent damage of the endothelial glycocalyx. Kidney Int. 2019, 95, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, M.; Andrieu, T.; Neuenschwander, S.; Bruggmann, R.; Mordasini, D.; Frey, F.J.; Vogt, B.; Frey, B.M. Regulation of 11beta-hydroxysteroid dehydrogenase type 2 by microRNA. Hypertension 2014, 64, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.; Kuppusamy, M.; Velarde-Miranda, C.; Gomez-Sanchez, C.M.; Plonczynski, M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. 11betaHSD2 Efficacy in Preventing Transcriptional Activation of the Mineralocorticoid Receptor by Corticosterone. J. Endocr. Soc. 2021, 5, bvab146. [Google Scholar] [CrossRef]
- Deuchar, G.A.; McLean, D.; Hadoke, P.W.; Brownstein, D.G.; Webb, D.J.; Mullins, J.J.; Chapman, K.; Seckl, J.R.; Kotelevtsev, Y.V. 11beta-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe−/− mice. Endocrinology 2011, 152, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Mullins, L.J.; Kenyon, C.J.; Bailey, M.A.; Conway, B.R.; Diaz, M.E.; Mullins, J.J. Mineralocorticoid Excess or Glucocorticoid Insufficiency: Renal and Metabolic Phenotypes in a Rat Hsd11b2 Knockout Model. Hypertension 2015, 66, 667–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Mladinov, D.; Pietrusz, J.L.; Usa, K.; Liang, M. Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc. Res. 2009, 81, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevtsev, Y.; Brown, R.W.; Fleming, S.; Kenyon, C.; Edwards, C.R.; Seckl, J.R.; Mullins, J.J. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J. Clin. Investig. 1999, 103, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Lovati, E.; Ferrari, P.; Dick, B.; Jostarndt, K.; Frey, B.M.; Frey, F.J.; Schorr, U.; Sharma, A.M. Molecular basis of human salt sensitivity: The role of the 11beta-hydroxysteroid dehydrogenase type 2. J. Clin. Endocrinol. Metab. 1999, 84, 3745–3749. [Google Scholar] [CrossRef] [Green Version]
- Lauterburg, M.; Escher, G.; Dick, B.; Ackermann, D.; Frey, F.J. Uninephrectomy reduces 11beta-hydroxysteroid dehydrogenase type 1 and type 2 concomitantly with an increase in blood pressure in rats. J. Endocrinol. 2012, 214, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Huesler, C.; Lauterburg, M.; Frey, B.M.; Frey, F.J. Evidence for glucocorticoid-mediated hypertension after uninephrectomy. Physiol. Rep. 2013, 1, e00101. [Google Scholar] [CrossRef]
- Quinkler, M.; Zehnder, D.; Lepenies, J.; Petrelli, M.D.; Moore, J.S.; Hughes, S.V.; Cockwell, P.; Hewison, M.; Stewart, P.M. Expression of renal 11beta-hydroxysteroid dehydrogenase type 2 is decreased in patients with impaired renal function. Eur. J. Endocrinol. 2005, 153, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Melcescu, E.; Phillips, J.; Moll, G.; Subauste, J.S.; Koch, C.A. 11Beta-hydroxylase deficiency and other syndromes of mineralocorticoid excess as a rare cause of endocrine hypertension. Horm. Metab. Res. 2012, 44, 867–878. [Google Scholar] [CrossRef]
- Alikhani-Koupaei, R.; Fouladkou, F.; Fustier, P.; Cenni, B.; Sharma, A.M.; Deter, H.C.; Frey, B.M.; Frey, F.J. Identification of polymorphisms in the human 11beta-hydroxysteroid dehydrogenase type 2 gene promoter: Functional characterization and relevance for salt sensitivity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3618–3628. [Google Scholar] [CrossRef]
- Williams, J.S. Evolving research in nongenomic actions of aldosterone. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 198–203. [Google Scholar] [CrossRef]
- Dooley, R.; Harvey, B.J.; Thomas, W. Non-genomic actions of aldosterone: From receptors and signals to membrane targets. Mol. Cell. Endocrinol. 2012, 350, 223–234. [Google Scholar] [CrossRef]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Munster, C.; Chraibi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef] [Green Version]
- Wildling, L.; Hinterdorfer, P.; Kusche-Vihrog, K.; Treffner, Y.; Oberleithner, H. Aldosterone receptor sites on plasma membrane of human vascular endothelium detected by a mechanical nanosensor. Pflug. Arch. 2009, 458, 223–230. [Google Scholar] [CrossRef]
- Ricchiuti, V.; Lapointe, N.; Pojoga, L.; Yao, T.; Tran, L.; Williams, G.H.; Adler, G.K. Dietary sodium intake regulates angiotensin II type 1, mineralocorticoid receptor, and associated signaling proteins in heart. J. Endocrinol. 2011, 211, 47–54. [Google Scholar] [CrossRef]
- Grossmann, C.; Husse, B.; Mildenberger, S.; Schreier, B.; Schuman, K.; Gekle, M. Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim. Biophys. Acta 2010, 1803, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Ashton, A.W.; Le, T.Y.; Gomez-Sanchez, C.E.; Morel-Kopp, M.C.; McWhinney, B.; Hudson, A.; Mihailidou, A.S. Role of Nongenomic Signaling Pathways Activated by Aldosterone during Cardiac Reperfusion Injury. Mol. Endocrinol. 2015, 29, 1144–1155. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Meyer, M.R. Nicolaus Copernicus and the rapid vascular responses to aldosterone. Trends Endocrinol. Metab. 2015, 26, 396–398. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2016, 7, 94–111. [Google Scholar] [CrossRef] [Green Version]
- Caroccia, B.; Seccia, T.M.; Piazza, M.; Prisco, S.; Zanin, S.; Iacobone, M.; Lenzini, L.; Pallafacchina, G.; Domening, O.; Poglitsch, M.; et al. Aldosterone Stimulates Its Biosynthesis Via a Novel GPER-Mediated Mechanism. J. Clin. Endocrinol. Metab. 2019, 104, 6316–6324. [Google Scholar] [CrossRef]
- Gros, R.; Ding, Q.; Liu, B.; Chorazyczewski, J.; Feldman, R.D. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am. J. Physiol. Cell Physiol. 2013, 304, C532–C540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, K.V.; McCurley, A.T.; Jaffe, I.Z. Direct contribution of vascular mineralocorticoid receptors to blood pressure regulation. Clin. Exp. Pharmacol. Physiol. 2013, 40, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mattia, R.A.; Mariangelo, J.I.E.; Blanco, P.G.; Jaquenod De Giusti, C.; Portiansky, E.L.; Mundina-Weilenmann, C.; Aiello, E.A.; Orlowski, A. The activation of the G protein-coupled estrogen receptor (GPER) prevents and regresses cardiac hypertrophy. Life Sci. 2020, 242, 117211. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.H.; Ferguson, J.K.; Burford, J.L.; Gevorgyan, H.; Nakano, D.; Harper, S.J.; Bates, D.O.; Peti-Peterdi, J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J. Am. Soc. Nephrol. 2012, 23, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2011, 462, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Mordi, I.; Tzemos, N. Is reversal of endothelial dysfunction still an attractive target in modern cardiology? World J. Cardiol. 2014, 6, 824–835. [Google Scholar] [CrossRef]
- McVeigh, G.E.; Brennan, G.M.; Johnston, G.D.; McDermott, B.J.; McGrath, L.T.; Henry, W.R.; Andrews, J.W.; Hayes, J.R. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992, 35, 771–776. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Adams, M.R.; Clarkson, P.; Robinson, J.; McCredie, R.; Donald, A.; Deanfield, J.E. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. N. Engl. J. Med. 1996, 334, 150–154. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Georgakopoulos, D.; Bull, C.; Thomas, O.; Robinson, J.; Deanfield, J.E. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation 1993, 88, 2149–2155. [Google Scholar] [CrossRef] [Green Version]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Nishizaka, M.K.; Zaman, M.A.; Green, S.A.; Renfroe, K.Y.; Calhoun, D.A. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004, 109, 2857–2861. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.J.; Biegelsen, E.S.; Eberhardt, R.T.; Kahn, D.F.; Kingwell, B.A.; Vita, J.A. Low-renin hypertension with relative aldosterone excess is associated with impaired NO-mediated vasodilation. Hypertension 2005, 46, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Demirkiran, A.; Everaars, H.; Elitok, A.; van de Ven, P.M.; Smulders, Y.M.; Dreijerink, K.M.; Tanakol, R.; Ozcan, M. Hypertension with primary aldosteronism is associated with increased carotid intima-media thickness and endothelial dysfunction. J. Clin. Hypertens. 2019, 21, 932–941. [Google Scholar] [CrossRef]
- Abiose, A.K.; Mansoor, G.A.; Barry, M.; Soucier, R.; Nair, C.K.; Hager, D. Effect of spironolactone on endothelial function in patients with congestive heart failure on conventional medical therapy. Am. J. Cardiol. 2004, 93, 1564–1566. [Google Scholar] [CrossRef]
- Nowak, K.L.; Gitomer, B.; Farmer-Bailey, H.; Wang, W.; Malaczewski, M.; Klawitter, J.; You, Z.; George, D.; Patel, N.; Jovanovich, A.; et al. Mineralocorticoid Antagonism and Vascular Function in Early Autosomal Dominant Polycystic Kidney Disease: A Randomized Controlled Trial. Am. J. Kidney Dis. 2019, 74, 213–223. [Google Scholar] [CrossRef]
- Zeng, Y.; Tarbell, J.M. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE 2014, 9, e86249. [Google Scholar] [CrossRef] [Green Version]
- Yen, W.; Cai, B.; Yang, J.; Zhang, L.; Zeng, M.; Tarbell, J.M.; Fu, B.M. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE 2015, 10, e0117133. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Simon, S.I.; Curry, F.R. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [Green Version]
- Ramnath, R.D.; Butler, M.J.; Newman, G.; Desideri, S.; Russell, A.; Lay, A.C.; Neal, C.R.; Qiu, Y.; Fawaz, S.; Onions, K.L.; et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 2020, 97, 951–965. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, T.; Beese, M.; Wyss, K.; Klinge, U.; Haller, H.; Haubitz, M.; Fiebeler, A. Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension 2013, 61, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberleithner, H.; Riethmuller, C.; Ludwig, T.; Shahin, V.; Stock, C.; Schwab, A.; Hausberg, M.; Kusche, K.; Schillers, H. Differential action of steroid hormones on human endothelium. J. Cell Sci. 2006, 119, 1926–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, A.H.; Satchell, S.C. Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J. Pathol. 2012, 226, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Ferguson, J.K.; Ramnath, R.D.; Onions, K.L.; Ogier, A.S.; Gamez, M.; Down, C.J.; Skinner, L.; Wong, K.H.; Dixon, L.K.; et al. Mineralocorticoid receptor antagonism in diabetes reduces albuminuria by preserving the glomerular endothelial glycocalyx. JCI Insight 2023, 8, e154164. [Google Scholar] [CrossRef]
- Satchell, S. The role of the glomerular endothelium in albumin handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef]
- Praga, M.; Morales, E. Obesity, proteinuria and progression of renal failure. Curr. Opin. Nephrol. Hypertens. 2006, 15, 481–486. [Google Scholar] [CrossRef]
- Bolignano, D.; Palmer, S.C.; Navaneethan, S.D.; Strippoli, G.F. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2014, 4, CD007004. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E. Summary of KDIGO 2012 CKD Guideline: Behind the scenes, need for guidance, and a framework for moving forward. Kidney Int. 2014, 85, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.K.; Szeto, C.C. Mineralocorticoid receptor antagonist for renal protection. Ren. Fail. 2012, 34, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, N.; Kawamura, T.; Okonogi, H.; Ishii, T.; Hosoya, T. The long-term antiproteinuric effect of eplerenone, a selective aldosterone blocker, in patients with non-diabetic chronic kidney disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2012, 13, 113–117. [Google Scholar] [CrossRef]
- Sengul, E.; Sahin, T.; Sevin, E.; Yilmaz, A. Effect of spironolactone on urinary protein excretion in patients with chronic kidney disease. Ren. Fail. 2009, 31, 928–932. [Google Scholar] [CrossRef]
- Schreier, B.; Rabe, S.; Schneider, B.; Ruhs, S.; Grossmann, C.; Hauptmann, S.; Blessing, M.; Neumann, J.; Gekle, M. Aldosterone/NaCl-induced renal and cardiac fibrosis is modulated by TGF-beta responsiveness of T cells. Hypertens. Res. 2011, 34, 623–629. [Google Scholar] [CrossRef]
- Piecha, G.; Koleganova, N.; Gross, M.L.; Geldyyev, A.; Adamczak, M.; Ritz, E. Regression of glomerulosclerosis in subtotally nephrectomized rats: Effects of monotherapy with losartan, spironolactone, and their combination. Am. J. Physiol. Ren. Physiol. 2008, 295, F137–F144. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Dhoopun, A.R.; Yuan, Y.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F520–F531. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.L.; Nikolic-Paterson, D.J.; Han, Y.; Ozols, E.; Ma, F.Y.; Young, M.J.; Tesch, G.H. Myeloid Mineralocorticoid Receptor Activation Contributes to Progressive Kidney Disease. J. Am. Soc. Nephrol. 2014, 25, 2231–2240. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Brennan, F.E.; Young, M.J.; Fuller, P.J.; Cole, T.J. A direct effect of aldosterone on endothelin-1 gene expression in vivo. Endocrinology 2007, 148, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Raina, R.; Chauvin, A.; Chakraborty, R.; Nair, N.; Shah, H.; Krishnappa, V.; Kusumi, K. The Role of Endothelin and Endothelin Antagonists in Chronic Kidney Disease. Kidney Dis. 2020, 6, 22–34. [Google Scholar] [CrossRef]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Grone, H.J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Krug, A.W.; Kopprasch, S.; Ziegler, C.G.; Dippong, S.; Catar, R.A.; Bornstein, S.R.; Morawietz, H.; Gekle, M. Aldosterone rapidly induces leukocyte adhesion to endothelial cells: A new link between aldosterone and arteriosclerosis? Hypertension 2007, 50, e156–e157. [Google Scholar] [CrossRef] [Green Version]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lother, A.; Furst, D.; Bergemann, S.; Gilsbach, R.; Grahammer, F.; Huber, T.B.; Hilgendorf, I.; Bode, C.; Moser, M.; Hein, L. Deoxycorticosterone Acetate/Salt-Induced Cardiac but not Renal Injury Is Mediated by Endothelial Mineralocorticoid Receptors Independently From Blood Pressure. Hypertension 2016, 67, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprio, M.; Newfell, B.G.; la Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive networks controlling vascular inflammation. Arter. Thromb. Vasc. Biol. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Riethmuller, C.; Schillers, H.; MacGregor, G.A.; de Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef] [Green Version]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Meinitzer, A.; Boehm, B.O.; Marz, W. Plasma aldosterone levels are associated with increased cardiovascular mortality: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Eur. Heart J. 2010, 31, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Ivanes, F.; Susen, S.; Mouquet, F.; Pigny, P.; Cuilleret, F.; Sautiere, K.; Collet, J.P.; Beygui, F.; Hennache, B.; Ennezat, P.V.; et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur. Heart J. 2012, 33, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Wentzel, J.J.; Chatzizisis, Y.S.; Gijsen, F.J.; Giannoglou, G.D.; Feldman, C.L.; Stone, P.H. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: Current understanding and remaining questions. Cardiovasc. Res. 2012, 96, 234–243. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, X.; Wang, Z.; Gao, X.; Ge, Z.; Gu, Y.; Ye, P.; Chao, Y.; Zhu, L.; Li, X.; et al. AMP-activated protein kinase regulates glycocalyx impairment and macrophage recruitment in response to low shear stress. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 7202–7212. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Haddad, N.; Rajan, J.; Nagaraja, H.N.; Agarwal, A.K.; Hebert, L.A. Usual ACE inhibitor therapy in CKD patients is associated with lower plasma aldosterone levels than usual angiotensin receptor blocker therapy. Kidney Blood Press. Res. 2007, 30, 299–305. [Google Scholar] [CrossRef]
- Schjoedt, K.J. The renin-angiotensin-aldosterone system and its blockade in diabetic nephropathy: Main focus on the role of aldosterone. Dan. Med. Bull. 2011, 58, B4265. [Google Scholar]
- Sato, A.; Saruta, T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. J. Int. Med. Res. 2001, 29, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Hayashi, K.; Naruse, M.; Saruta, T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003, 41, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.; Farmer, C.K.; Irving, J.; Stevens, P.E. Acute kidney injury: An acceptable risk of treatment with renin-angiotensin system blockade in primary care? Can. J. Kidney Health Dis. 2015, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Struthers, A.D.; Fahey, T.; Watson, A.D.; Macdonald, T.M. Spironolactone use and renal toxicity: Population based longitudinal analysis. BMJ 2010, 340, c1768. [Google Scholar] [CrossRef] [Green Version]
- Sica, D.A. Pharmacokinetics and pharmacodynamics of mineralocorticoid blocking agents and their effects on potassium homeostasis. Heart Fail. Rev. 2005, 10, 23–29. [Google Scholar] [CrossRef]
- Hill, N.R.; Lasserson, D.; Thompson, B.; Perera-Salazar, R.; Wolstenholme, J.; Bower, P.; Blakeman, T.; Fitzmaurice, D.; Little, P.; Feder, G.; et al. Benefits of Aldosterone Receptor Antagonism in Chronic Kidney Disease (BARACK D) trial-a multi-centre, prospective, randomised, open, blinded end-point, 36-month study of 2616 patients within primary care with stage 3b chronic kidney disease to compare the efficacy of spironolactone 25 mg once daily in addition to routine care on mortality and cardiovascular outcomes versus routine care alone: Study protocol for a randomized controlled trial. Trials 2014, 15, 160. [Google Scholar] [CrossRef] [Green Version]
- Deinum, J.; Riksen, N.P.; Lenders, J.W. Pharmacological treatment of aldosterone excess. Pharm. Ther. 2015, 154, 120–133. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Kolkhof, P.; Delbeck, M.; Kretschmer, A.; Steinke, W.; Hartmann, E.; Barfacker, L.; Eitner, F.; Albrecht-Kupper, B.; Schafer, S. Finerenone, a novel selective nonsteroidal mineralocorticoid receptor antagonist protects from rat cardiorenal injury. J. Cardiovasc. Pharmacol. 2014, 64, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Snelder, N.; Heinig, R.; Drenth, H.J.; Joseph, A.; Kolkhof, P.; Lippert, J.; Garmann, D.; Ploeger, B.; Eissing, T. Population Pharmacokinetic and Exposure-Response Analysis of Finerenone: Insights Based on Phase IIb Data and Simulations to Support Dose Selection for Pivotal Trials in Type 2 Diabetes with Chronic Kidney Disease. Clin. Pharm. 2020, 59, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; et al. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J. Am. Soc. Nephrol. 2022, 33, 225–237. [Google Scholar] [CrossRef]
- Pivonello, R.; Fleseriu, M.; Newell-Price, J.; Bertagna, X.; Findling, J.; Shimatsu, A.; Gu, F.; Auchus, R.; Leelawattana, R.; Lee, E.J.; et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): A multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020, 8, 748–761. [Google Scholar] [CrossRef]
- Cai, T.Q.; Stribling, S.; Tong, X.; Xu, L.; Wisniewski, T.; Fontenot, J.A.; Struthers, M.; Akinsanya, K.O. Rhesus monkey model for concurrent analyses of in vivo selectivity, pharmacokinetics and pharmacodynamics of aldosterone synthase inhibitors. J. Pharm. Toxicol. Methods 2015, 71, 137–146. [Google Scholar] [CrossRef]
- Papillon, J.P.; Lou, C.; Singh, A.K.; Adams, C.M.; Ksander, G.M.; Beil, M.E.; Chen, W.; Leung-Chu, J.; Fu, F.; Gan, L.; et al. Discovery of N-[5-(6-Chloro-3-cyano-1-methyl-1H-indol-2-yl)-pyridin-3-ylmethyl]-ethanesulfonam ide, a Cortisol-Sparing CYP11B2 Inhibitor that Lowers Aldosterone in Human Subjects. J. Med. Chem. 2015, 58, 9382–9394. [Google Scholar] [CrossRef]
- Sakakibara, R.; Sasaki, W.; Onda, Y.; Yamaguchi, M.; Ushirogochi, H.; Hiraga, Y.; Sato, K.; Nishio, M.; Egi, Y.; Takedomi, K.; et al. Discovery of Novel Pyrazole-Based Selective Aldosterone Synthase (CYP11B2) Inhibitors: A New Template to Coordinate the Heme-Iron Motif of CYP11B2. J. Med. Chem. 2018, 61, 5594–5608. [Google Scholar] [CrossRef]
- Sparks, S.M.; Danger, D.P.; Hoekstra, W.J.; Leesnitzer, T.; Schotzinger, R.J.; Yates, C.M.; Becherer, J.D. Development of Highly Selective Pyrimidine-Based Aldosterone Synthase (CYP11B2) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1056–1060. [Google Scholar] [CrossRef]
- Freeman, M.W.; Halvorsen, Y.D.; Marshall, W.; Pater, M.; Isaacsohn, J.; Pearce, C.; Murphy, B.; Alp, N.; Srivastava, A.; Bhatt, D.L.; et al. Phase 2 Trial of Baxdrostat for Treatment-Resistant Hypertension. N. Engl. J. Med. 2023, 388, 395–405. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crompton, M.; Skinner, L.J.; Satchell, S.C.; Butler, M.J. Aldosterone: Essential for Life but Damaging to the Vascular Endothelium. Biomolecules 2023, 13, 1004. https://doi.org/10.3390/biom13061004
Crompton M, Skinner LJ, Satchell SC, Butler MJ. Aldosterone: Essential for Life but Damaging to the Vascular Endothelium. Biomolecules. 2023; 13(6):1004. https://doi.org/10.3390/biom13061004
Chicago/Turabian StyleCrompton, Michael, Laura J. Skinner, Simon C. Satchell, and Matthew J. Butler. 2023. "Aldosterone: Essential for Life but Damaging to the Vascular Endothelium" Biomolecules 13, no. 6: 1004. https://doi.org/10.3390/biom13061004
APA StyleCrompton, M., Skinner, L. J., Satchell, S. C., & Butler, M. J. (2023). Aldosterone: Essential for Life but Damaging to the Vascular Endothelium. Biomolecules, 13(6), 1004. https://doi.org/10.3390/biom13061004