
CC-Chemokine Receptor-2 Expression in Osteoblasts Contributes to Cartilage and Bone Damage during Post-Traumatic Osteoarthritis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Antibodies
2.2. Animals
2.3. Induction of OA
2.4. Behavioral Studies
2.5. Histopathologic Assessment of Arthritis
2.6. Osteophyte Assessment
2.7. Immunohistochemistry (IHC) Studies
2.8. Assessment of Synovial Thickness
2.9. Statistical Analysis
3. Results
3.1. Ccr2 Inactivation in Bone Tissue
3.2. Early Osteoblast-Ccr2 Inactivation Decreases Cartilage Damage Induced by Injury
3.3. Early Osteoblast-Ccr2 Inactivation before Injury Reduces Bone Thickness but Does Not Affect Osteophyte Formation
3.4. Early Osteoblast-Ccr2 Inactivation Does Not Affect Synovial Hyperplasia Induced by Injury
3.5. Late Osteoblast-Ccr2 Inactivation during OA Progression Decreases Cartilage Damage Induced by DMM
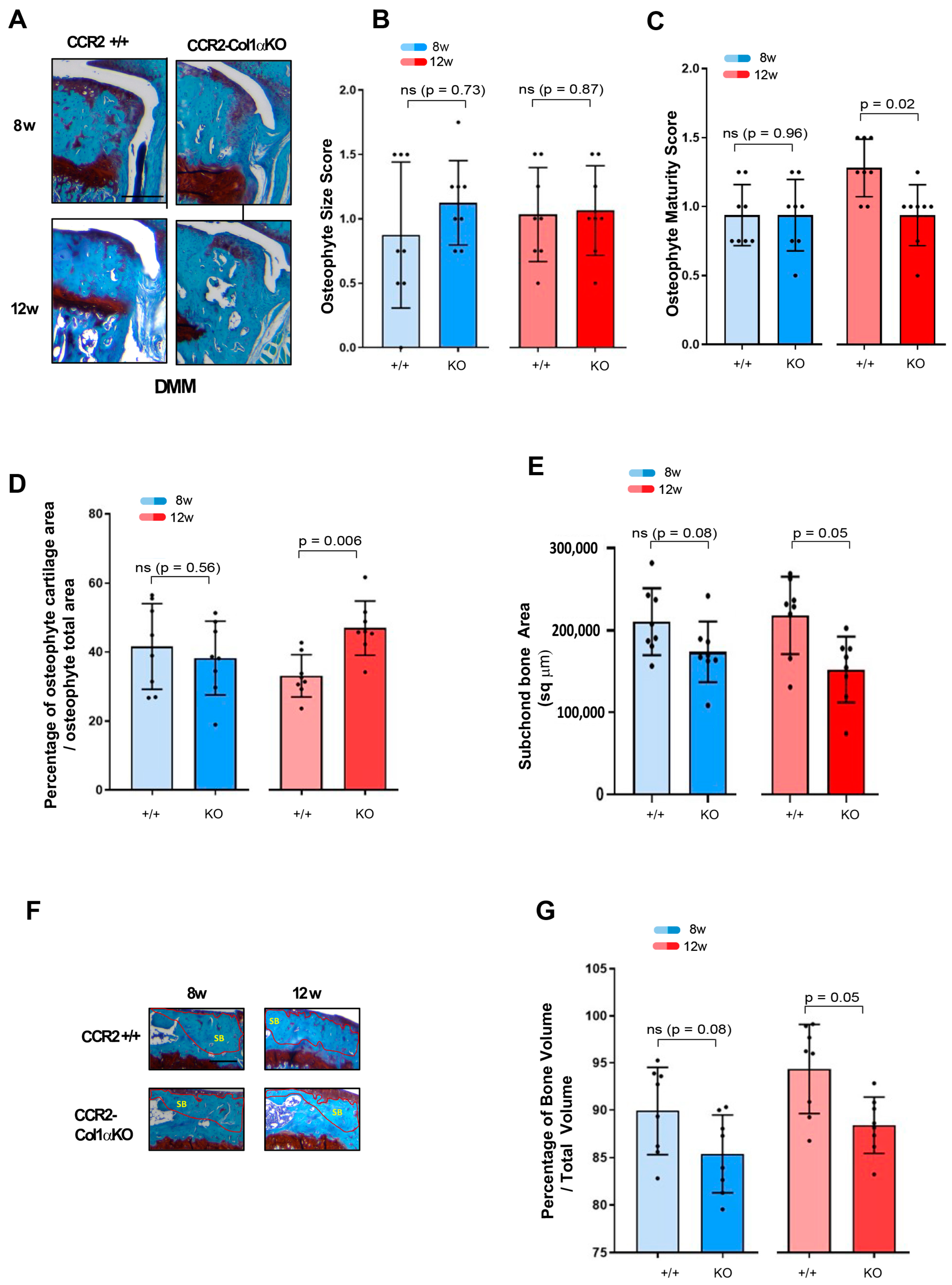
3.6. Late Osteoblast-Ccr2 Inactivation during OA Progression Delays Bone Thickness but Does Not Affect Osteophyte Formation
3.7. Late Osteoblast-Ccr2 Inactivation Does Not Affect Synovial Hyperplasia Induced by Injury
3.8. Reduced PTOA Joint Damage following Osteoblast-CCR2 Inactivation Diminishes Pain Responses at the Severe Stage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Supplemental Methods
References
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Lepus, C.M.; Wang, Q.; Wong, H.H.; Lingampalli, N.; Oliviero, F.; Punzi, L.; Giori, N.J.; Goodman, S.B.; Chu, C.R.; et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 2017, 76, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Appleton, C.T.; Usmani, S.E.; Pest, M.A.; Pitelka, V.; Mort, J.S.; Beier, F. Reduction in disease progression by inhibition of transforming growth factor alpha-CCL2 signaling in experimental posttraumatic osteoarthritis. Arthritis Rheumatol. 2015, 67, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, B.E. Serum and synovial fluid chemokine ligand 2/monocyte chemoattractant protein 1 concentrations correlates with symptomatic severity in patients with knee osteoarthritis. Ann. Clin. Biochem. 2015, 52, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, L.; Jordan, J.M.; Shi, X.A.; Renner, J.B.; Schwartz, T.A.; Nelson, A.E.; Barrow, D.A.; Kraus, V.B.; Spagnoli, A. Associations between the chemokine biomarker CCL2 and knee osteoarthritis outcomes: The Johnston County Osteoarthritis Project. Osteoarthr. Cartil. 2018, 26, 1257–1261. [Google Scholar] [CrossRef]
- Longobardi, L.; Temple, J.D.; Tagliafierro, L.; Willcockson, H.; Esposito, A.; D’Onofrio, N.; Stein, E.; Li, T.; Myers, T.J.; Ozkan, H.; et al. Role of the C-C chemokine receptor-2 in a murine model of injury-induced osteoarthritis. Osteoarthr. Cartil. 2017, 25, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Tran, P.B.; Das, R.; Ghoreishi-Haack, N.; Ren, D.; Miller, R.J.; Malfait, A.M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 20602–20607. [Google Scholar] [CrossRef]
- Miotla Zarebska, J.; Chanalaris, A.; Driscoll, C.; Burleigh, A.; Miller, R.E.; Malfait, A.M.; Stott, B.; Vincent, T.L. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthr. Cartil. 2017, 25, 406–412. [Google Scholar] [CrossRef]
- Ozkan, H.; Di Francesco, M.; Willcockson, H.; Valdes-Fernandez, J.; Di Francesco, V.; Granero-Molto, F.; Prosper, F.; Decuzzi, P.; Longobardi, L. Sustained inhibition of CC-chemokine receptor-2 via intraarticular deposition of polymeric microplates in post-traumatic osteoarthritis. Drug Deliv. Transl. Res. 2022, 13, 689–701. [Google Scholar] [CrossRef]
- Willcockson, H.; Ozkan, H.; Arbeeva, L.; Mucahit, E.; Musawwir, L.; Longobardi, L. Early Ablation of Ccr2 in Aggrecan-expressing cells Following Knee Injury Ameliorates Joint Damage and Pain during Post-traumatic Osteoarthritis. Osteoarthr. Cartil. 2022, 30, 1616–1630. [Google Scholar] [CrossRef]
- Binder, N.B.; Niederreiter, B.; Hoffmann, O.; Stange, R.; Pap, T.; Stulnig, T.M.; Mack, M.; Erben, R.G.; Smolen, J.S.; Redlich, K. Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat. Med. 2009, 15, 417–424. [Google Scholar] [CrossRef]
- Xing, Z.; Lu, C.; Hu, D.; Yu, Y.Y.; Wang, X.; Colnot, C.; Nakamura, M.; Wu, Y.; Miclau, T.; Marcucio, R.S. Multiple roles for CCR2 during fracture healing. Dis. Model. Mech. 2010, 3, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Quinones, M.P.; Ahuja, S.K.; Jimenez, F.; Schaefer, J.; Garavito, E.; Rao, A.; Chenaux, G.; Reddick, R.L.; Kuziel, W.A.; Ahuja, S.S. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J. Clin. Investig. 2004, 113, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.R.D.A.; Andrade, I.; Queiroz-Junior, C.; Garlet, T.P.; Garlet, G.; Cunha, F.Q.; Teixeira, M.M.; da Silva, T.A. Role of CCR2 in orthodontic tooth movement. Am. J. Orthod. Dentofac. Orthop. 2012, 141, 153–160. [Google Scholar] [CrossRef] [PubMed]
- von Luettichau, I.; Segerer, S.; Wechselberger, A.; Notohamiprodjo, M.; Nathrath, M.; Kremer, M.; Henger, A.; Djafarzadeh, R.; Burdach, S.; Huss, R.; et al. A complex pattern of chemokine receptor expression is seen in osteosarcoma. BMC Cancer 2008, 8, 23. [Google Scholar] [CrossRef]
- Hopwood, B.; Tsykin, A.; Findlay, D.M.; Fazzalari, N.L. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res. Ther. 2007, 9, R100. [Google Scholar] [CrossRef]
- Kim, J.E.; Nakashima, K.; de Crombrugghe, B. Transgenic mice expressing a ligand-inducible cre recombinase in osteoblasts and odontoblasts: A new tool to examine physiology and disease of postnatal bone and tooth. Am. J. Pathol. 2004, 165, 1875–1882. [Google Scholar] [CrossRef]
- Willenborg, S.; Lucas, T.; van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.A.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Kanki, K.; Wang, E.; Peluso, D.; et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheum. 2004, 50, 2547–2558. [Google Scholar] [CrossRef]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.L.; Blanchet, T.J.; Peluso, D.; Hopkins, B.; Morris, E.A.; Glasson, S.S. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthr. Cartil. 2007, 15, 695–700. [Google Scholar] [CrossRef]
- Bove, S.E.; Calcaterra, S.L.; Brooker, R.M.; Huber, C.M.; Guzman, R.E.; Juneau, P.L.; Schrier, D.J.; Kilgore, K.S. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthr. Cartil./OARS Osteoarthr. Res. Soc. 2003, 11, 821–830. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef] [PubMed]
- McNulty, M.A.; Loeser, R.F.; Davey, C.; Callahan, M.F.; Ferguson, C.M.; Carlson, C.S. A Comprehensive Histological Assessment of Osteoarthritis Lesions in Mice. Cartilage 2011, 2, 354–363. [Google Scholar] [CrossRef]
- Nagira, K.; Ikuta, Y.; Shinohara, M.; Sanada, Y.; Omoto, T.; Kanaya, H.; Nakasa, T.; Ishikawa, M.; Adachi, N.; Miyaki, S.; et al. Histological scoring system for subchondral bone changes in murine models of joint aging and osteoarthritis. Sci. Rep. 2020, 10, 10077. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Rowe, M.A.; Harper, L.R.; McNulty, M.A.; Lau, A.G.; Carlson, C.S.; Leng, L.; Bucala, R.J.; Miller, R.A.; Loeser, R.F. Reduced Osteoarthritis Severity in Aged Mice With Deletion of Macrophage Migration Inhibitory Factor. Arthritis Rheumatol. 2017, 69, 352–361. [Google Scholar] [CrossRef]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Fan, T.; Chen, S.; Zeng, M.; Li, J.; Wang, X.; Ruan, G.; Cao, P.; Zhang, Y.; Chen, T.; Ou, Q.; et al. Osteophytes mediate the associations between cartilage morphology and changes in knee symptoms in patients with knee osteoarthritis. Arthritis Res. Ther. 2022, 24, 217. [Google Scholar] [CrossRef]
- Eraltan, H.; Cacina, C.; Kahraman, O.T.; Kurt, O.; Aydogan, H.Y.; Uyar, M.; Can, A.; Cakmakoglu, B. MCP-1 and CCR2 gene variants and the risk for osteoporosis and osteopenia. Genet. Test. Mol. Biomark. 2012, 16, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liu, M.; Bennett, S.; Wang, Z.; Pfleger, K.D.G.; Xu, J. The molecular structure and role of CCL2 (MCP-1) and C-C chemokine receptor CCR2 in skeletal biology and diseases. J. Cell. Physiol. 2021, 236, 7211–7222. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef] [PubMed]
- Madry, H.; van Dijk, C.N.; Mueller-Gerbl, M. The basic science of the subchondral bone. Knee Surg. Sport. Traumatol. Arthrosc. 2010, 18, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, S.; Roman-Blas, J.A.; Largo, R.; Herrero-Beaumont, G. Subchondral bone as a key target for osteoarthritis treatment. Biochem. Pharmacol. 2012, 83, 315–323. [Google Scholar] [CrossRef]
- Fang, H.; Huang, L.; Welch, I.; Norley, C.; Holdsworth, D.W.; Beier, F.; Cai, D. Early Changes of Articular Cartilage and Subchondral Bone in The DMM Mouse Model of Osteoarthritis. Sci. Rep. 2018, 8, 2855. [Google Scholar] [CrossRef]
- Malfait, A.M.; Schnitzer, T.J. Towards a mechanism-based approach to pain management in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 654–664. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Woolf, A.D.; Pfleger, B. Burden of major musculoskeletal conditions. Bull. World Health Organ. 2003, 81, 646–656. [Google Scholar]
- Sun, Q.; Li, G.; Liu, D.; Xie, W.; Xiao, W.; Li, Y.; Cai, M. Peripheral nerves in the tibial subchondral bone: The role of pain and homeostasis in osteoarthritis. Bone Jt. Res. 2022, 11, 439–452. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Investig. 2019, 129, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.; Gill, S.E.; de Camin, S.M.; Wilson, D.; McWilliams, D.F.; Walsh, D.A. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.L.; Geha, P.Y.; Baliki, M.N.; Katz, J.; Schnitzer, T.J.; Apkarian, A.V. Brain activity for chronic knee osteoarthritis: Dissociating evoked pain from spontaneous pain. Eur. J. Pain 2011, 15, 843 e841-814. [Google Scholar] [CrossRef]
- Abbadie, C.; Lindia, J.A.; Cumiskey, A.M.; Peterson, L.B.; Mudgett, J.S.; Bayne, E.K.; DeMartino, J.A.; MacIntyre, D.E.; Forrest, M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. USA 2003, 100, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Peat, G.; Thomas, E.; Hay, E.; McCall, I.; Croft, P. Symptoms and radiographic osteoarthritis: Not as discordant as they are made out to be? Ann. Rheum. Dis. 2007, 66, 86–91. [Google Scholar] [CrossRef]
- Neogi, T.; Felson, D.; Niu, J.; Nevitt, M.; Lewis, C.E.; Aliabadi, P.; Sack, B.; Torner, J.; Bradley, L.; Zhang, Y. Association between radiographic features of knee osteoarthritis and pain: Results from two cohort studies. BMJ 2009, 339, b2844. [Google Scholar] [CrossRef]
- Miller, R.E.; Malfait, A.M. Can we target CCR2 to treat osteoarthritis? The trick is in the timing! Osteoarthr. Cartil. 2017, 25, 799–801. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OA Parameters | Week 2 | Week 4 | |||||||
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| ACS | DMM CCR2+/+ | 3.01 (0.01) | 12.13 | 2 | 2.06 (0.69) | −2.23 [0.03] | 5.81 | 4.25 | 3.91 (1.17) |
| DMM CCR2-AggKO | 4.88 | 0.75 | 0.81 (0.42) | 11.19 | 5.75 | 5.34 (1.38) | |||
| Saf-O | DMM CCR2+/+ | 3.34 (0.002) | 12.5 | 3 | 2.97 (0.51) | −0.32 [0.75] | 8.06 | 4.25 | 4.53 (1.27) |
| DMM CCR2-AggKO | 4.5 | 1.25 | 1.06 (0.55) | 8.94 | 4.75 | 4.75 (1.11) | |||
| Cartilage Histomorphometry | DMM CCR2+/+ | −0.05 (0.96) | 8.38 | 30,842 | 29,817 (4810) | 1.84 (0.13) | 10.75 | 29,506 | 29,551 (7452) |
| DMM CCR2-AggKO | 8.63 | 29,873 | 31,073 (7051) | 6.25 | 21,135 | 21,992 (5516) | |||
| OA Parameters | Week 8 | Week 12 | |||||||
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| ACS | DMM CCR2+/+ | 0.47 (0.64) | 9.13 | 7.63 | 7.75 (0.97) | 3.01 (0.01) | 12.13 | 11.5 | 11.16 (0.77) |
| DMM CCR2-AggKO | 7.88 | 7.5 | 7.13 (1.76) | 4.88 | 7.25 | 6.78 (3.29) | |||
| Saf-O | DMM CCR2+/+ | 2.00 (0.06) | 10.94 | 8.38 | 7.97 (1.38) | 3.33 (0.002) | 12.5 | 11.63 | 11.47 (0.65) |
| DMM CCR2-AggKO | 6.06 | 6.63 | 6.42 (1.62) | 4.5 | 6.13 | 6.03 (2.18) | |||
| Cartilage Histomorphometry | DMM CCR2+/+ | −0.05 (0.96) | 8.38 | 18,963 | 18,161 (6042) | −2.99 (0.01) | 4.88 | 7835 | 8369 (2508) |
| DMM CCR2-AggKO | 8.63 | 18,218 | 18,364 (9115) | 12.13 | 20,436 | 22,937 (10,655) | |||
| OA Parameters | Week 2 | Week 4 | |||||||
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| Osteophyte Cartilage Quantification | DMM CCR2+/+ | −2.26 [0.08] | 5.75 | 34.4 | 35.72 (13.59) | 0–0.37 [0.71] | 8 | 52.98 | 49.12 (21.50) |
| DMM CCR2-AggKO | 11.25 | 51.51 | 52.34 (13.70) | 9 | 60.19 | 53.87 (23.88) | |||
| Osteophyte Size | DMM CCR2+/+ | 0.73 (0.61) | 9.38 | 1 | 1.00 (0.19) | 0.43 (0.66) | 9.06 | 0.88 | 0.94 (0.26) |
| DMM CCR2-AggKO | 7.63 | 1 | 0.91 (0.33) | 7.94 | 0.88 | 0.88 (0.38) | |||
| Osteophyte Maturity | DMM CCR2+/+ | 0.91 (0.36) | 9.63 | 1 | 1.03 (0.45) | 0.65 (0.51) | 9.31 | 1 | 1.06 (0.50) |
| DMM CCR2-AggKO | 7.38 | 0.88 | 0.81 (0.46) | 7.69 | 0.88 | 0.84 (0.33) | |||
| %BV over TV | DMM CCR2+/+ | 1.42 (0.21) | 10.25 | 85.76 | 84.76 (3.03) | 0.79 (0.43) | 9.5 | 88.3 | 88.16 (5.38) |
| DMM CCR2-AggKO | 6.75 | 80.64 | 80.59 (6.61) | 7.5 | 86.74 | 85.54 (3.81) | |||
| Subchondral Bone Thickness | DMM CCR2+/+ | 0–0.26 [0.87] | 8.13 | 94,856 | 103,601 (21,658) | −0.16 [0.87] | 8.25 | 130,217 | 136,746 (36,572) |
| DMM CCR2-AggKO | 8.88 | 105,288 | 119,710 (47,463) | 8.75 | 127,770 | 138,730 (32,706) | |||
| OA Parameters | Week 8 | Week 12 | |||||||
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| Osteophyte Cartilage Quantification | DMM CCR2+/+ | 0.47 (0.71) | 9.13 | 42.42 | 43.20 (10.90) | −0.68 [0.71] | 7.63 | 32.13 | 31.86 (15.99) |
| DMM CCR2-AggKO | 7.88 | 34.9 | 39.61 (13.39) | 9.38 | 38.17 | 35.77 (11.86) | |||
| Osteophyte Size | DMM CCR2+/+ | 1.66 (0.20) | 10.5 | 1.63 | 1.69 (0.62) | 2.43 (0.08) | 11.38 | 1.63 | 1.78 (0.41) |
| DMM CCR2-AggKO | 6.5 | 1.25 | 1.22 (0.25) | 5.63 | 1.25 | 1.22 (0.36) | |||
| Osteophyte Maturity | DMM CCR2+/+ | 1.07 (0.28) | 9.81 | 1.38 | 1.53 (0.82) | 1.81 (0.07) | 10.69 | 1.63 | 1.69 (0.32) |
| DMM CCR2-AggKO | 7.19 | 1 | 1.06 (0.32) | 6.31 | 1.13 | 1.28 (0.43) | |||
| %BV over TV | DMM CCR2+/+ | 1.63 (0.20) | 10.5 | 92.54 | 92.27 (4.24) | 3.20 (0.004) | 12.38 | 97.87 | 97.06 (3.08) |
| DMM CCR2-AggKO | 6.5 | 88.39 | 87.67 (5.25) | 4.63 | 84.99 | 84.81 (5.55) | |||
| Subchondral Bone Thickness | DMM CCR2+/+ | −1 (0.64) | 7.25 | 132,176 | 138,215 (26,144) | 3.10 (0.008) | 12.25 | 205,757 | 205,979 (36,612) |
| DMM CCR2-AggKO | 9.75 | 159,877 | 154,085 (28,876) | 4.75 | 113,190 | 118,064 (19,106) | |||
| OA Parameters | Week 8 | Week 12 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| ACS | CCR2+/+ | −0.21 [0.83] | 8.19 | 5.75 | 6.56 (2.58) | 2.79 (0.01) | 11.88 | 9.38 | 9.66 (1.63) |
| CCR2-AggKO | 8.81 | 6.38 | 6.47 (1.28) | 5.13 | 5.5 | 5.53 (2.51) | |||
| Saf-O | CCR2+/+ | 0 [1.00] | 8.5 | 7.5 | 7.56 (3.32) | 2.88 [0.01] | 10.79 | 10 | 9.75 (1.22) |
| CCR2-AggKO | 8.5 | 7.5 | 7.44 (0.75) | 4.21 | 7 | 6.36 (1.63) | |||
| Cartilage Histomorphometry | CCR2+/+ | −0.16 (0.87) | 8.25 | 21,074 | 23,807 (10,260) | −1.11 (0.54) | 7.13 | 20,086 | 19,973 (3049) |
| CCR2-AggKO | 8.75 | 26,515 | 24,075 (8390) | 9.88 | 24,930 | 25,629 (7527) | |||
| OA Parameters | Week 8 | Week 12 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | Z (p-Value) | Mean Score (Rank Sums) | Median | Mean (SD) | ||
| Osteophyte Cartilage Quantification | CCR2+/+ | 0.58 [0.56] | 9.25 | 41.93 | 41.62 (12.42) | −2.99 [0.006] | 4.88 | 31.98 | 33.05 (6.13) |
| CCR2-AggKO | 7.75 | 38.43 | 38.27 (10.71) | 12.13 | 45.78 | 46.90 (7.85) | |||
| Osteophyte Size | CCR2+/+ | −0.91 [0.73] | 7.38 | 0.75 | 0.88 (0.57) | −0.16 [0.87] | 8.25 | 1 | 1.03 (0.36) |
| CCR2-AggKO | 9.63 | 1.13 | 1.13 (0.33) | 8.75 | 1 | 1.06 (0.35) | |||
| Osteophyte Maturity | CCR2+/+ | −0.06 [0.96] | 8.38 | 0.88 | 0.94 (0.22) | 2.55 [0.02] | 11.44 | 1.25 | 1.28 (0.21) |
| CCR2-AggKO | 8.63 | 1 | 0.94 (0.26) | 5.56 | 1 | 0.94 (0.22) | |||
| %BV over TV | CCR2+/+ | 1.73 [0.08] | 10.63 | 91.03 | 89.93 (4.61) | 2.26 [0.05] | 11.25 | 96.1 | 94.34 (4.73) |
| CCR2-AggKO | 6.38 | 85.61 | 85.39 (4.08) | 5.75 | 88.37 | 88.4 (2.96) | |||
| Subchondral Bone Thickness | CCR2+/+ | 1.73 [0.08] | 10.63 | 199,278 | 210,692 (40,762) | 2.26 [0.05] | 11.25 | 23,0354 | 21,8213 (47,127) |
| CCR2-AggKO | 6.38 | 168,541 | 173,802 (37,031) | 5.75 | 161,005 | 152,247 (40,161) | |||
| Pain | Incapacitance Meter | Von-Frey Filaments | ||
|---|---|---|---|---|
| Time PO/Stage | DMM CCR2+/+ vs. DMM CCR2-Col1KO | DMM CCR2+/+ vs. DMM CCR2-Col1KO | DMM CCR2+/+ vs. Contralateral CCR2+/+ | DMM CCR2-Col1KO vs. Contralateral CCR2-Col1KO |
| 4 wks/mild | 0.01 (−0.04, 0.06) [p = 0.7480] | −0.33 (−1.03,0.37) [p = 0.3613] | 0.04 (−0.56,0.64) [p = 0.9132] | 0.23 (−0.45,0.91) [p = 0.5194] |
| 8 wks/moderate | 0.14 (0.03, 0.26) [p = 0.0177] | −0.19 (−0.88,0.51) [p = 0.6109] | 0.02 (−0.59,0.64) [p = 0.9455] | 0.21 (−0.46,0.88) [p = 0.5523] |
| 12 wks/severe | 0.06 (−0.01, 0.14) [p = 0.0974] | −0.46 (−0.95,0.03) [p = 0.0627] | −0.66 (−1.14, −0.19) [p = 0.0064] | 0 (−0.45,0.45) [p = 0.9912] |
| 16 wks/severe | 0.15 (0.04, 0.26) [p = 0.0097] | −0.02 (−0.4,0.36) [p = 0.9301] | −0.44 (−0.86,−0.02) [p = 0.0376] | −0.49 (−0.91, −0.07) [p = 0.0230] |
| 20 wks/severe | 0.16 (0.07, 0.24) [p = 0.0006] | −0.16 (−0.54,0.22) [p = 0.4156] | −0.29 (−0.65,0.08) [p = 0.1245] | 0 (−0.33,0.33) [p = 0.9872] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willcockson, H.; Ozkan, H.; Valdés-Fernández, J.; Arbeeva, L.; Mucahit, E.; Musawwir, L.; Hooper, L.B.; Granero-Moltó, F.; Prósper, F.; Longobardi, L. CC-Chemokine Receptor-2 Expression in Osteoblasts Contributes to Cartilage and Bone Damage during Post-Traumatic Osteoarthritis. Biomolecules 2023, 13, 891. https://doi.org/10.3390/biom13060891
Willcockson H, Ozkan H, Valdés-Fernández J, Arbeeva L, Mucahit E, Musawwir L, Hooper LB, Granero-Moltó F, Prósper F, Longobardi L. CC-Chemokine Receptor-2 Expression in Osteoblasts Contributes to Cartilage and Bone Damage during Post-Traumatic Osteoarthritis. Biomolecules. 2023; 13(6):891. https://doi.org/10.3390/biom13060891
Chicago/Turabian StyleWillcockson, Helen, Huseyin Ozkan, José Valdés-Fernández, Liubov Arbeeva, Esra Mucahit, Layla Musawwir, Lola B. Hooper, Froilán Granero-Moltó, Felipe Prósper, and Lara Longobardi. 2023. "CC-Chemokine Receptor-2 Expression in Osteoblasts Contributes to Cartilage and Bone Damage during Post-Traumatic Osteoarthritis" Biomolecules 13, no. 6: 891. https://doi.org/10.3390/biom13060891
APA StyleWillcockson, H., Ozkan, H., Valdés-Fernández, J., Arbeeva, L., Mucahit, E., Musawwir, L., Hooper, L. B., Granero-Moltó, F., Prósper, F., & Longobardi, L. (2023). CC-Chemokine Receptor-2 Expression in Osteoblasts Contributes to Cartilage and Bone Damage during Post-Traumatic Osteoarthritis. Biomolecules, 13(6), 891. https://doi.org/10.3390/biom13060891