



Insulin-Degrading Enzyme Interacts with Mitochondrial Ribosomes and Respiratory Chain Proteins
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
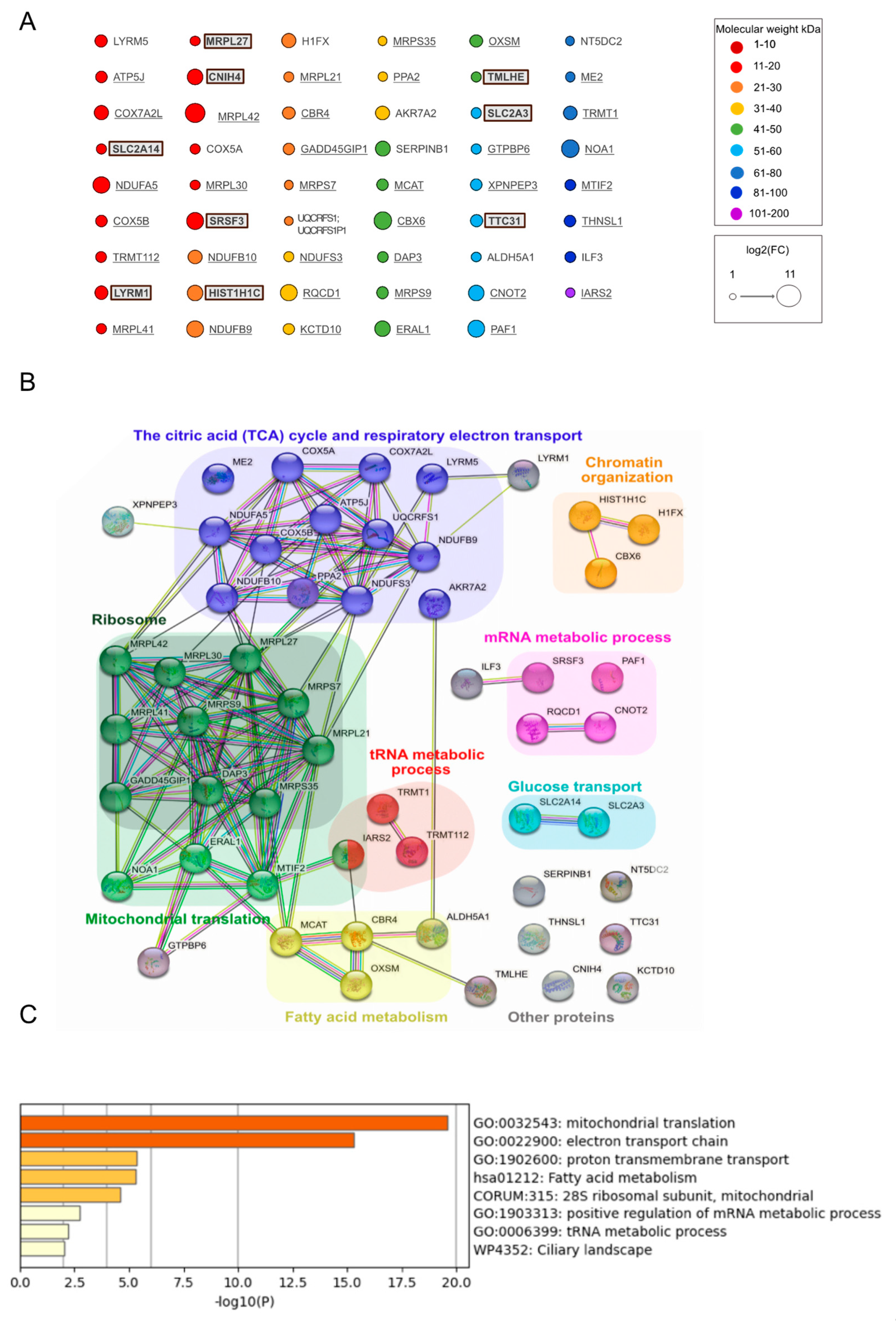{kind=link}
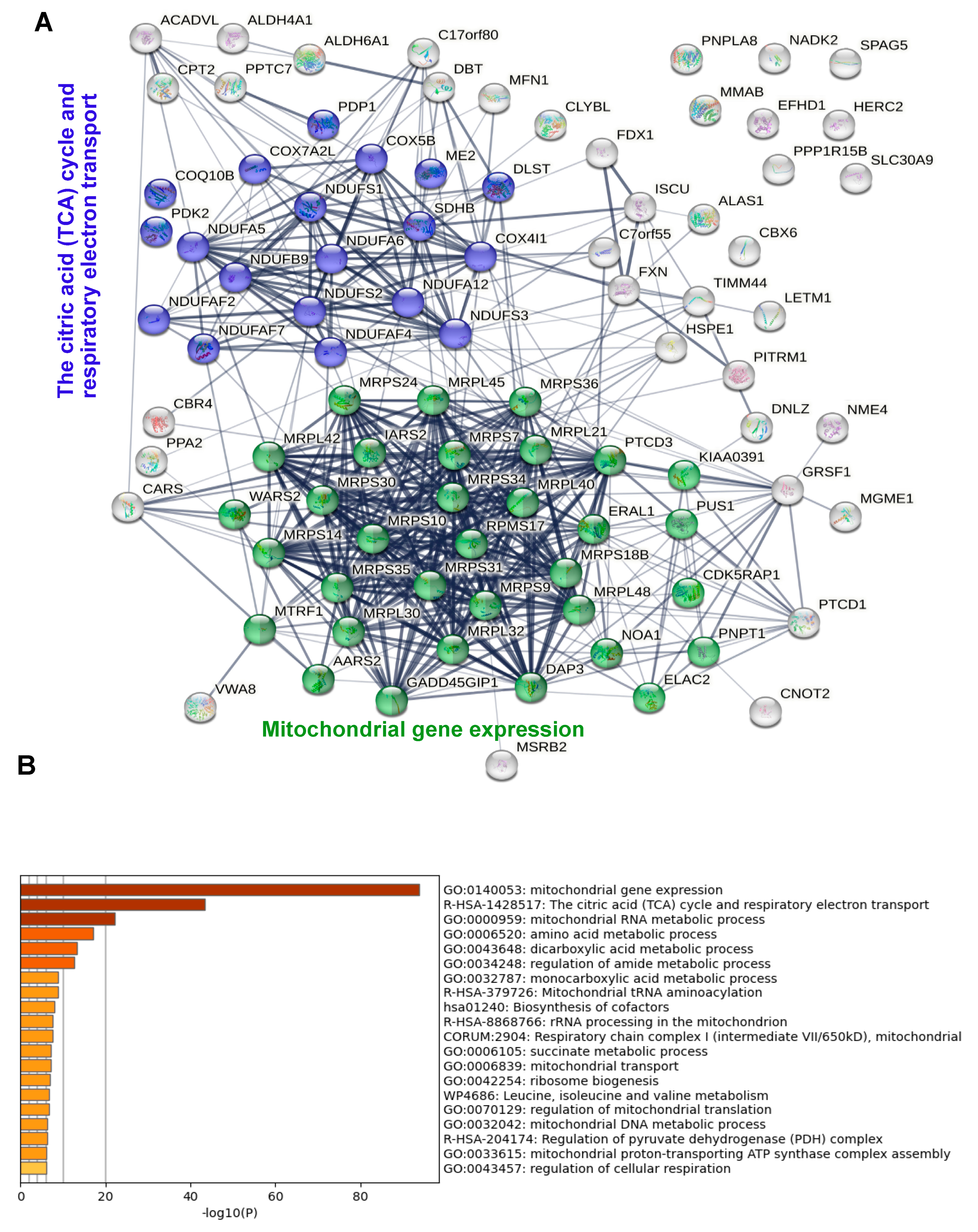{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of TurboID (Biotin Ligase BirA Mutant) Fusion Constructs, Lentivirus Production and Transduction
2.2. Confocal Microscopy
2.3. Western Blot
2.4. TurboID-Based Enzymatic Protein Labeling and Extraction of Biotinylated Proteins for Proteomic Analysis
2.5. Identification and Quantification of Proteins by NanoLC-MS/MS
2.6. Data Processing after LC-MS/MS Acquisition
2.7. IDE Interactome Data Analysis
3. Results
3.1. Design and Verification of the Proximity Biotinylation System
3.1.1. Constructs and Protein Expression
3.1.2. Protein Localization
3.1.3. Biotinylation Setup
3.2. Identification of Proteins Interacting with Wt and Mutant Mitochondrial IDE
3.3. Differential Control Analysis Corroborates Mitochondrial IDE Interactome and Reveals Potential Cytosolic IDE Interactomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.-J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef]
- Grasso, G.; Rizzarelli, E.; Spoto, G. How the binding and degrading capabilities of insulin degrading enzyme are affected by ubiquitin. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 1122–1126. [Google Scholar] [CrossRef]
- Saric, T.; Müller, D.; Seitz, H.-J.; Pavelic, K. Non-covalent interaction of ubiquitin with insulin-degrading enzyme. Mol. Cell. Endocrinol. 2003, 204, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ralat, L.A.; Kalas, V.; Zheng, Z.; Goldman, R.D.; Sosnick, T.R.; Tang, W.-J. Ubiquitin Is a Novel Substrate for Human Insulin-Degrading Enzyme. J. Mol. Biol. 2011, 406, 454–466. [Google Scholar] [CrossRef] [PubMed]
- McCord, L.A.; Liang, W.G.; Dowdell, E.; Kalas, V.; Hoey, R.J.; Koide, A.; Koide, S.; Tang, W.-J. Conformational states and recognition of amyloidogenic peptides of human insulin-degrading enzyme. Proc. Natl. Acad. Sci. USA 2013, 110, 13827–13832. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Hulse, R.E.; Tang, W.J. Amyloid beta-degrading cryptidases: Insulin degrading enzyme, presequence peptidase, and neprilysin. Cell Mol. Life Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.-J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulou, M.; Guo, Q.; Malito, E.; Schilling, A.B.; Tang, W.-J. Molecular Basis of Catalytic Chamber-assisted Unfolding and Cleavage of Human Insulin by Human Insulin-degrading Enzyme. J. Biol. Chem. 2009, 284, 14177–14188. [Google Scholar] [CrossRef]
- Noinaj, N.; Bhasin, S.K.; Song, E.S.; Scoggin, K.E.; Juliano, M.A.; Juliano, L.; Hersh, L.B.; Rodgers, D.W. Identification of the Allosteric Regulatory Site of Insulysin. PLoS ONE 2011, 6, e20864. [Google Scholar] [CrossRef]
- Im, H.; Manolopoulou, M.; Malito, E.; Shen, Y.; Zhao, J.; Neant-Fery, M.; Sun, C.-Y.; Meredith, S.C.; Sisodia, S.S.; Leissring, M.A.; et al. Structure of Substrate-free Human Insulin-degrading Enzyme (IDE) and Biophysical Analysis of ATP-induced Conformational Switch of IDE. J. Biol. Chem. 2007, 282, 25453–25463. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Deprez-Poulain, R.; Hennuyer, N.; Bosc, D.; Liang, W.G.; Enée, E.; Marechal, X.; Charton, J.; Totobenazara, J.; Berte, G.; Jahklal, J.; et al. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat. Commun. 2015, 6, 8250. [Google Scholar] [CrossRef] [PubMed]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-degrading enzyme (IDE): A novel heat shock-like protein. J. Biol. Chem. 2013, 288, 2281–2299. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Jackson, W.; de Souza, G.A.; Haarr, L.; Grose, C. Insulin-degrading enzyme binds to the nonglycosylated precursor of varicella-zoster virus gE protein found in the endoplasmic reticulum. J. Virol. 2010, 84, 847–855. [Google Scholar] [CrossRef]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin Degrading Enzyme Is a Cellular Receptor Mediating Varicella-Zoster Virus Infection and Cell-to-Cell Spread. Cell 2006, 127, 305–316. [Google Scholar] [CrossRef]
- Sbardella, D.; Tundo, G.R.; Sciandra, F.; Bozzi, M.; Gioia, M.; Ciaccio, C.; Tarantino, U.; Brancaccio, A.; Coletta, M.; Marini, S. Proteasome Activity Is Affected by Fluctuations in Insulin-Degrading Enzyme Distribution. PLoS ONE 2015, 10, e0132455. [Google Scholar] [CrossRef]
- Sbardella, D.; Tundo, G.R.; Coletta, A.; Marcoux, J.; Koufogeorgou, E.I.; Ciaccio, C.; Santoro, A.M.; Milardi, D.; Grasso, G.; Cozza, P.; et al. The insulin-degrading enzyme is an allosteric modulator of the 20S proteasome and a potential competitor of the 19S. Cell. Mol. Life Sci. 2018, 75, 3441–3456. [Google Scholar] [CrossRef]
- Schmitz, A.; Schneider, A.; Kummer, M.P.; Herzog, V. Endoplasmic Reticulum-Localized Amyloid beta-Peptide is Degraded in the Cytosol by Two Distinct Degradation Pathways. Traffic 2003, 5, 89–101. [Google Scholar] [CrossRef]
- Leissring, M.A.; Farris, W.; Wu, X.; Christodoulou, D.C.; Haigis, M.C.; Guarente, L.; Selkoe, D.J. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem. J. 2004, 383, 439–446. [Google Scholar] [CrossRef]
- Zhao, F.; Zou, M.-H. Role of the Mitochondrial Protein Import Machinery and Protein Processing in Heart Disease. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Wijaya, J.; Wei, H.; Noble, A.J.; Mancl, J.M.; Mo, S.; Lee, D.; King, J.V.L.; Pan, M.; Liu, C.; et al. Structural basis for the mechanisms of human presequence protease conformational switch and substrate recognition. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Z.; Ren, M.; Yang, X.; Liu, B.; Qi, H.; Yu, M.; Song, S.; Chen, S.; Liu, L.; et al. SIRT4 regulates PTEN stability through IDE in response to cellular stresses. FASEB J. 2019, 33, 5535–5547. [Google Scholar] [CrossRef]
- Hansson, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Beuzelin, C.; Evnouchidou, I.; Rigolet, P.; Cauvet-Burgevin, A.; Girard, P.-M.; Dardalhon, D.; Culina, S.; Gdoura, A.; van Endert, P.; Francesconi, S. Deletion of the Fission Yeast Homologue of Human Insulinase Reveals a TORC1-Dependent Pathway Mediating Resistance to Proteotoxic Stress. PLoS ONE 2013, 8, e67705. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Udeshi, N.D.; Myers, S.A.; Carr, S.A.; Ting, A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020, 15, 3971–3999. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Mixed Dimers of Insulin-degrading Enzyme Reveal a Cis Activation Mechanism. J. Biol. Chem. 2011, 286, 13852–13858. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, Y.; Tsuji, J.; Fu, S.-C.; Tomii, K.; Horton, P.; Imai, K. MitoFates: Improved Prediction of Mitochondrial Targeting Sequences and Their Cleavage Sites*. Mol. Cell. Proteom. 2015, 14, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Bruciaferri, N.; Tartari, G.; Martelli, P.L.; Casadio, R. DeepMito: Accurate prediction of protein sub-mitochondrial localization using convolutional neural networks. Bioinformatics 2019, 36, 56–64. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple functions of insulin-degrading enzyme: A metabolic crosslight? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, A.; Guerrera, C.; Waeckel-Énée, E.; Lipecka, J.; Bertocci, B.; van Endert, P. Insulin-Degrading Enzyme Interacts with Mitochondrial Ribosomes and Respiratory Chain Proteins. Biomolecules 2023, 13, 890. https://doi.org/10.3390/biom13060890
Yilmaz A, Guerrera C, Waeckel-Énée E, Lipecka J, Bertocci B, van Endert P. Insulin-Degrading Enzyme Interacts with Mitochondrial Ribosomes and Respiratory Chain Proteins. Biomolecules. 2023; 13(6):890. https://doi.org/10.3390/biom13060890
Chicago/Turabian StyleYilmaz, Ayse, Chiara Guerrera, Emmanuelle Waeckel-Énée, Joanna Lipecka, Barbara Bertocci, and Peter van Endert. 2023. "Insulin-Degrading Enzyme Interacts with Mitochondrial Ribosomes and Respiratory Chain Proteins" Biomolecules 13, no. 6: 890. https://doi.org/10.3390/biom13060890
APA StyleYilmaz, A., Guerrera, C., Waeckel-Énée, E., Lipecka, J., Bertocci, B., & van Endert, P. (2023). Insulin-Degrading Enzyme Interacts with Mitochondrial Ribosomes and Respiratory Chain Proteins. Biomolecules, 13(6), 890. https://doi.org/10.3390/biom13060890