
Assessing the Resilience of Machine Learning Classification Algorithms on SARS-CoV-2 Genome Sequences Generated with Long-Read Specific Errors



Abstract
:1. Introduction
2. Dataset and Methodology
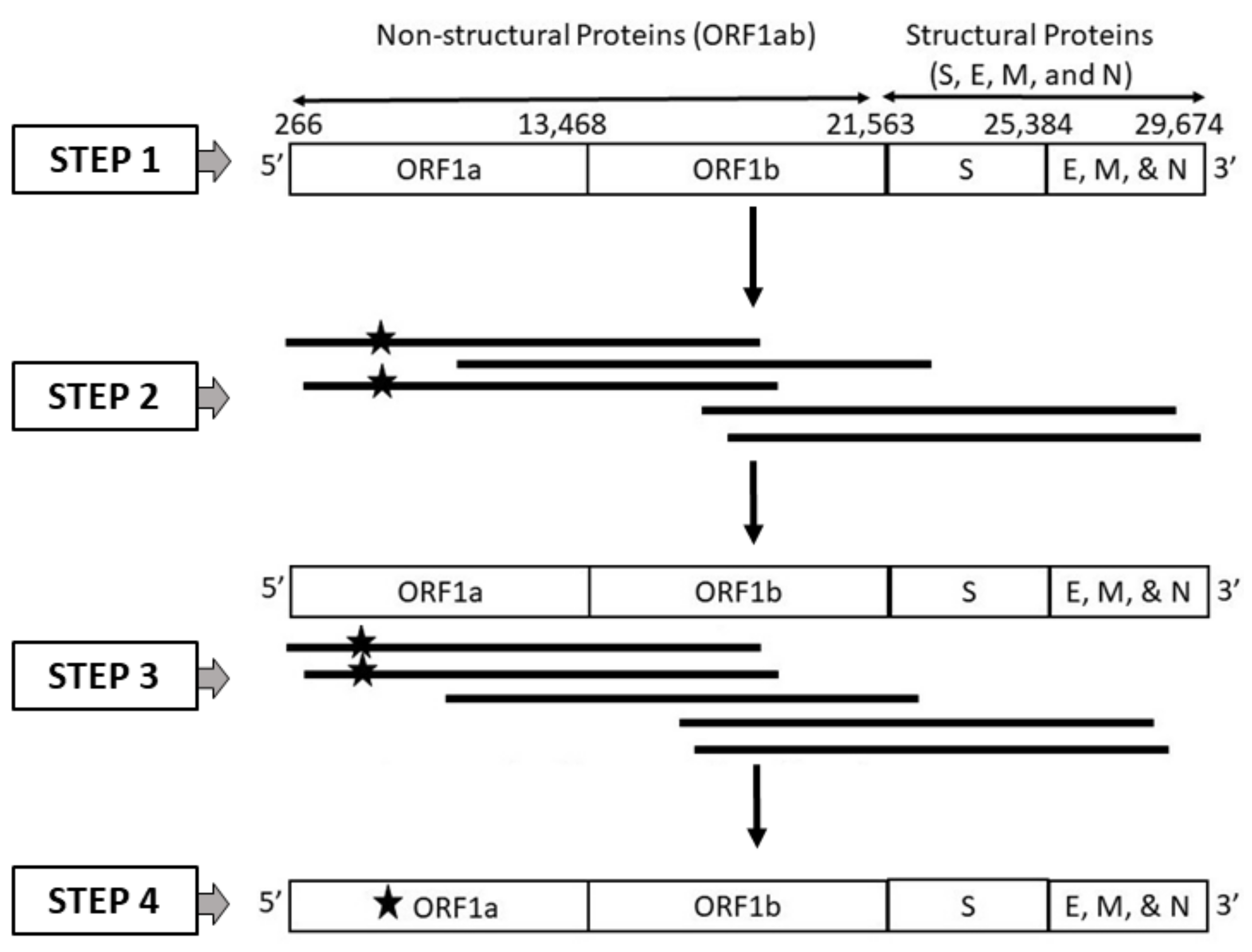
2.1. Dataset Generation
2.1.1. Dataset 1: High-Quality SARS-CoV-2 Genome Sequences
2.1.2. Dataset 2: SARS-CoV-2 Genome Sequences Generated from Long Reads Incorporating PacBio Sequencing Errors
2.1.3. Dataset 3: SARS-CoV-2 Genome Sequences Generated from Long Reads Incorporating Oxford Nanopore Technology (ONT) Sequencing Errors
2.1.4. Dataset 4: SARS-CoV-2 Genome Sequences Generated from Long Reads Incorporating Random Errors
2.2. Embedding Generation Methods
2.2.1. One-Hot Encoding (OHE)
2.2.2. Wasserstein-Distance-Based Generative Adversarial Network for Representation Learning (WDGRL)
2.2.3. String Kernel
2.2.4. Spaced k-Mers
2.2.5. Weighted k-Mers
2.2.6. The Weighted Position Weight Matrix (PWM)
2.3. Machine Learning Classification Algorithms: SVM, NB, MLP, KNN, RF, LR, and DT
2.3.1. Approach 1: Accuracy
2.3.2. Approach 2: Robustness
2.4. Data Visualization
3. Results and Discussion
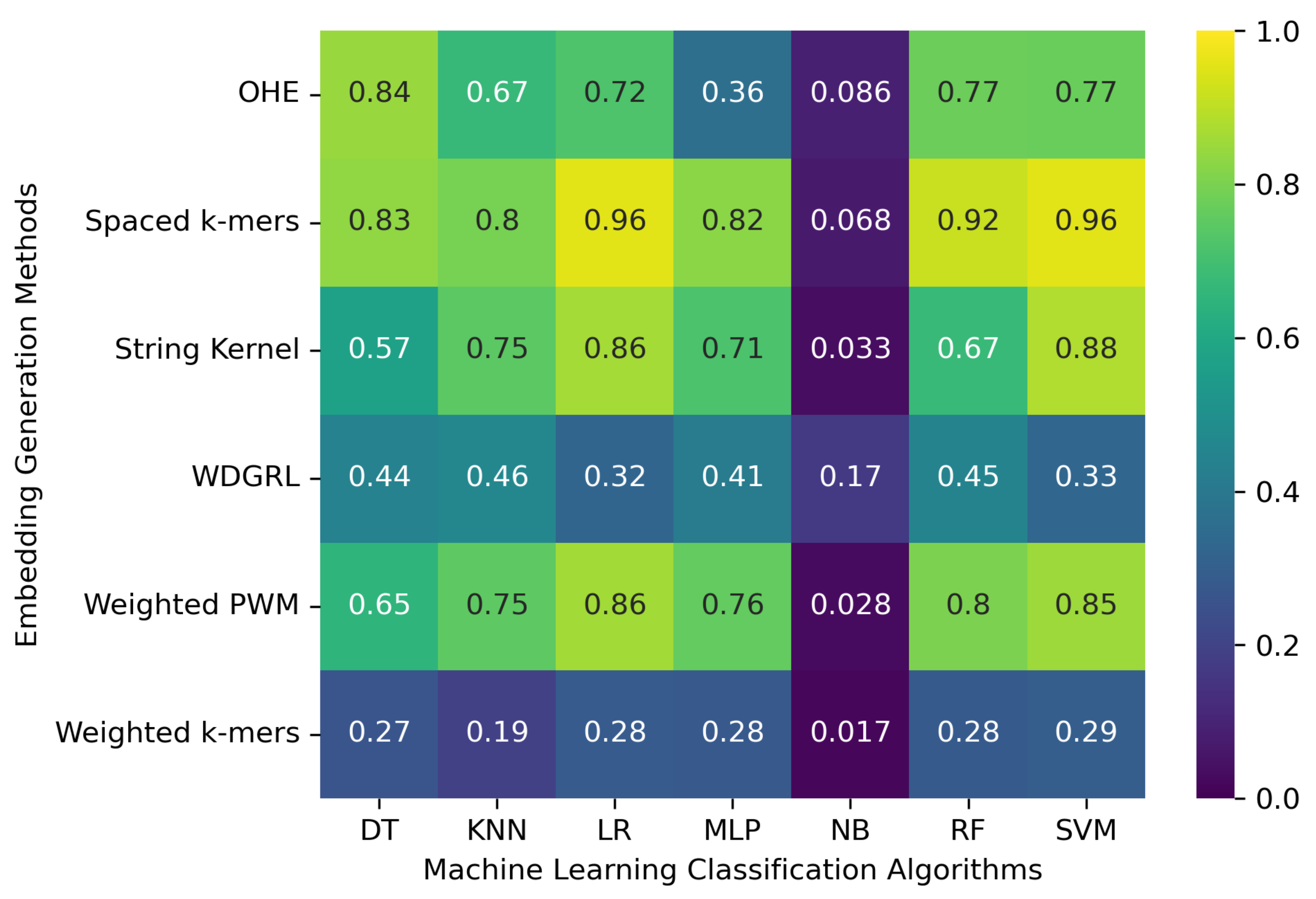
3.1. Accuracy Evaluation of Machine Learning Classification Algorithms Using Different Embedding Methods
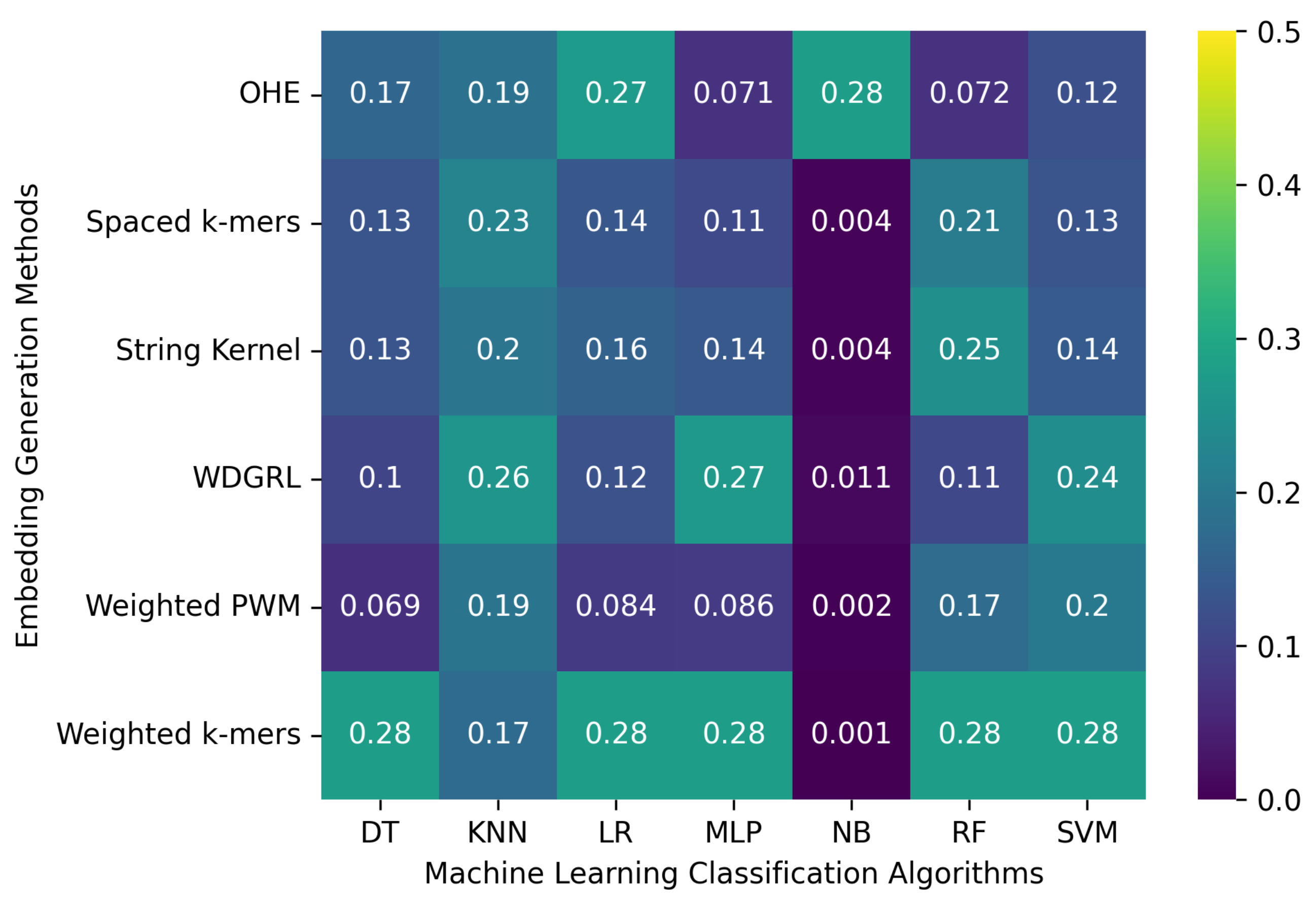
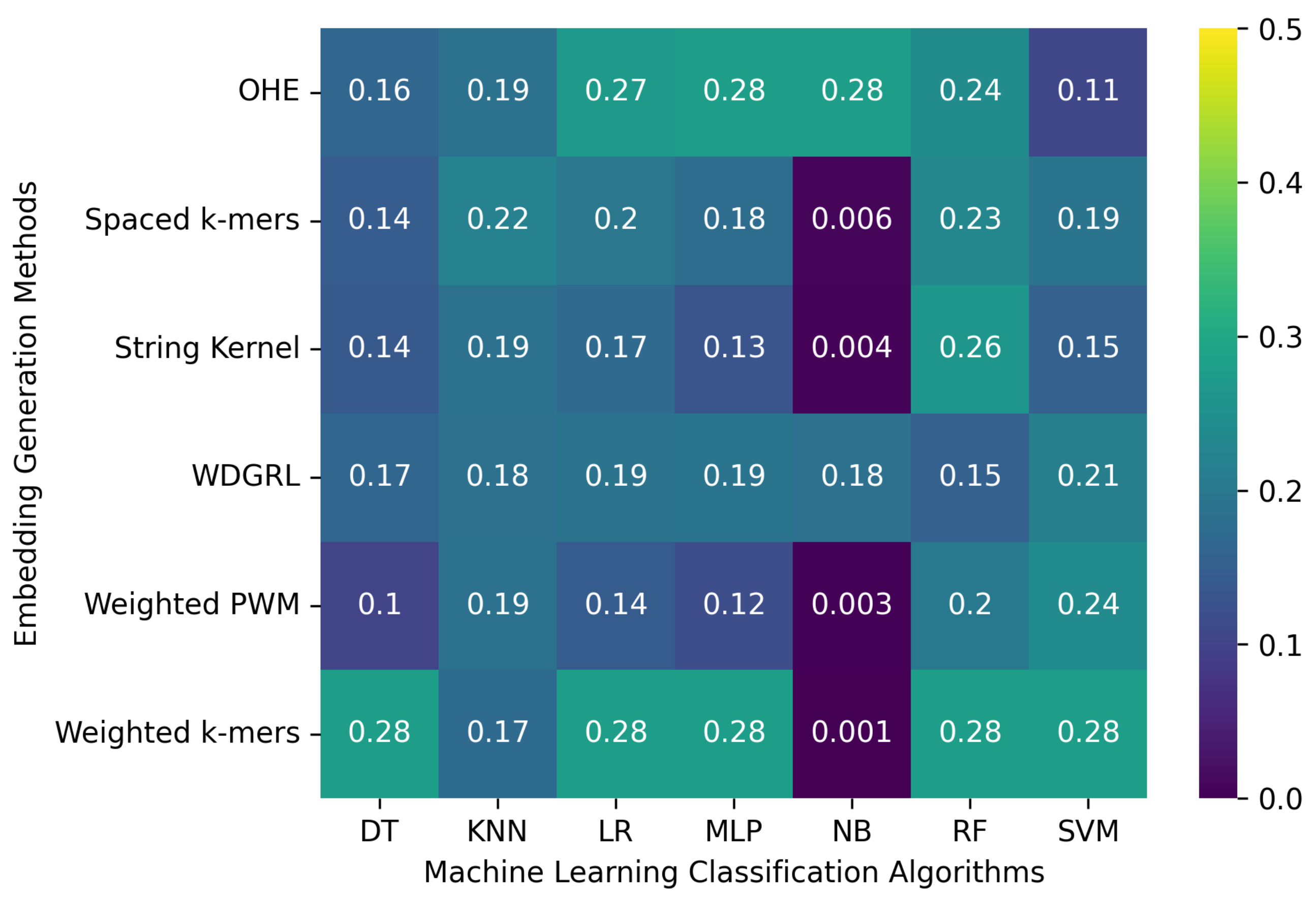
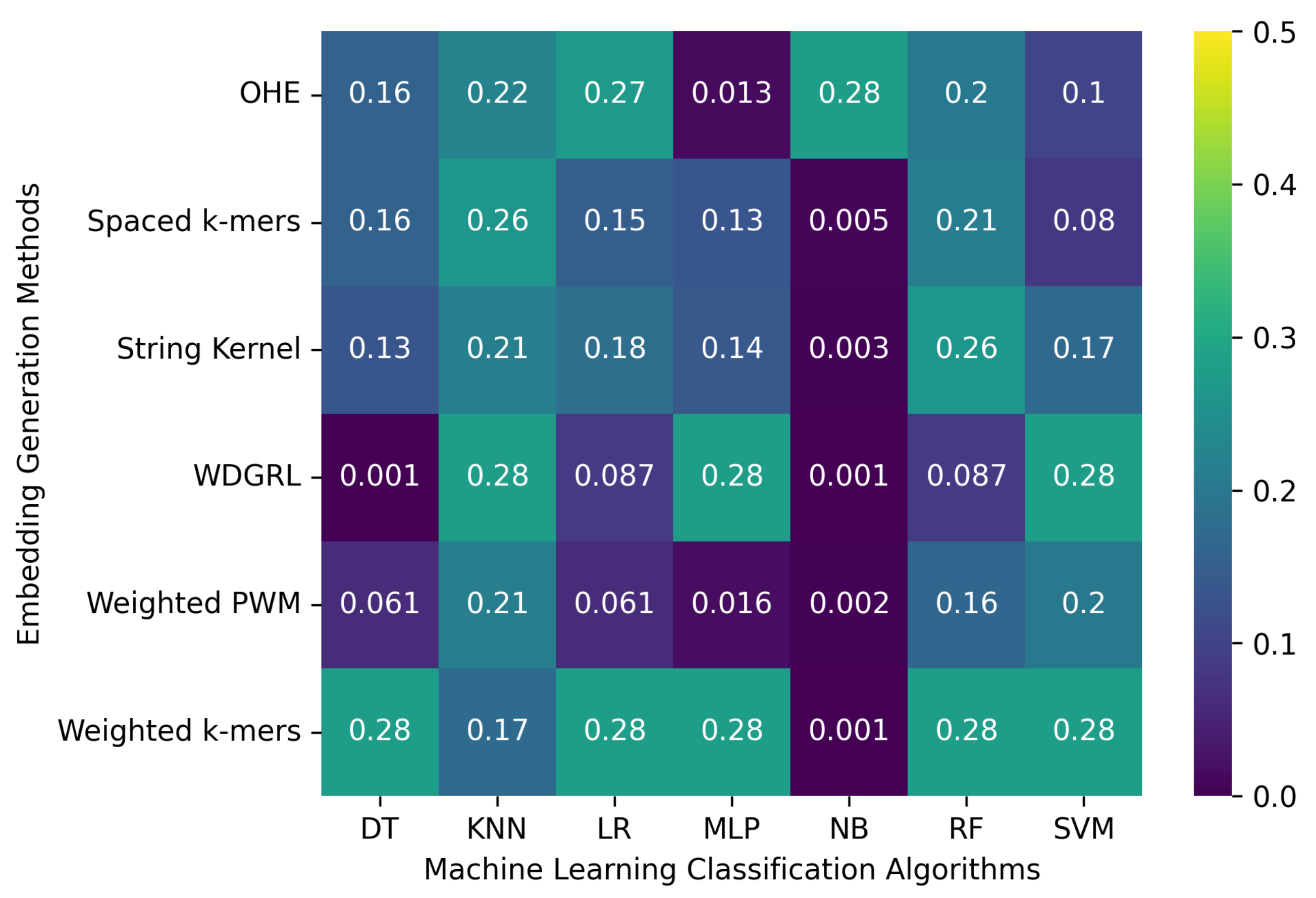
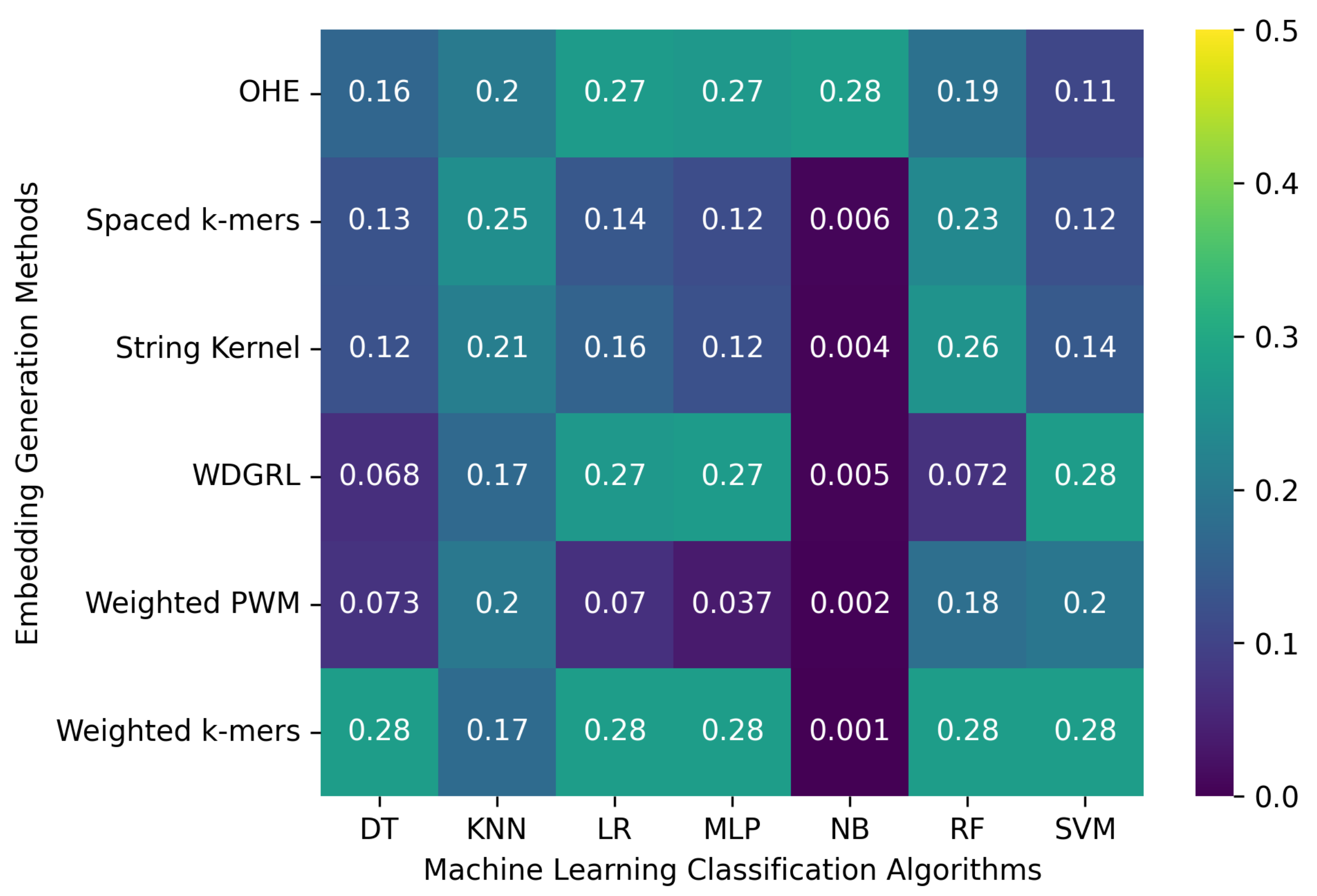
3.2. Robustness Evaluation of Machine Learning Classification Algorithms Using Different Embedding Methods
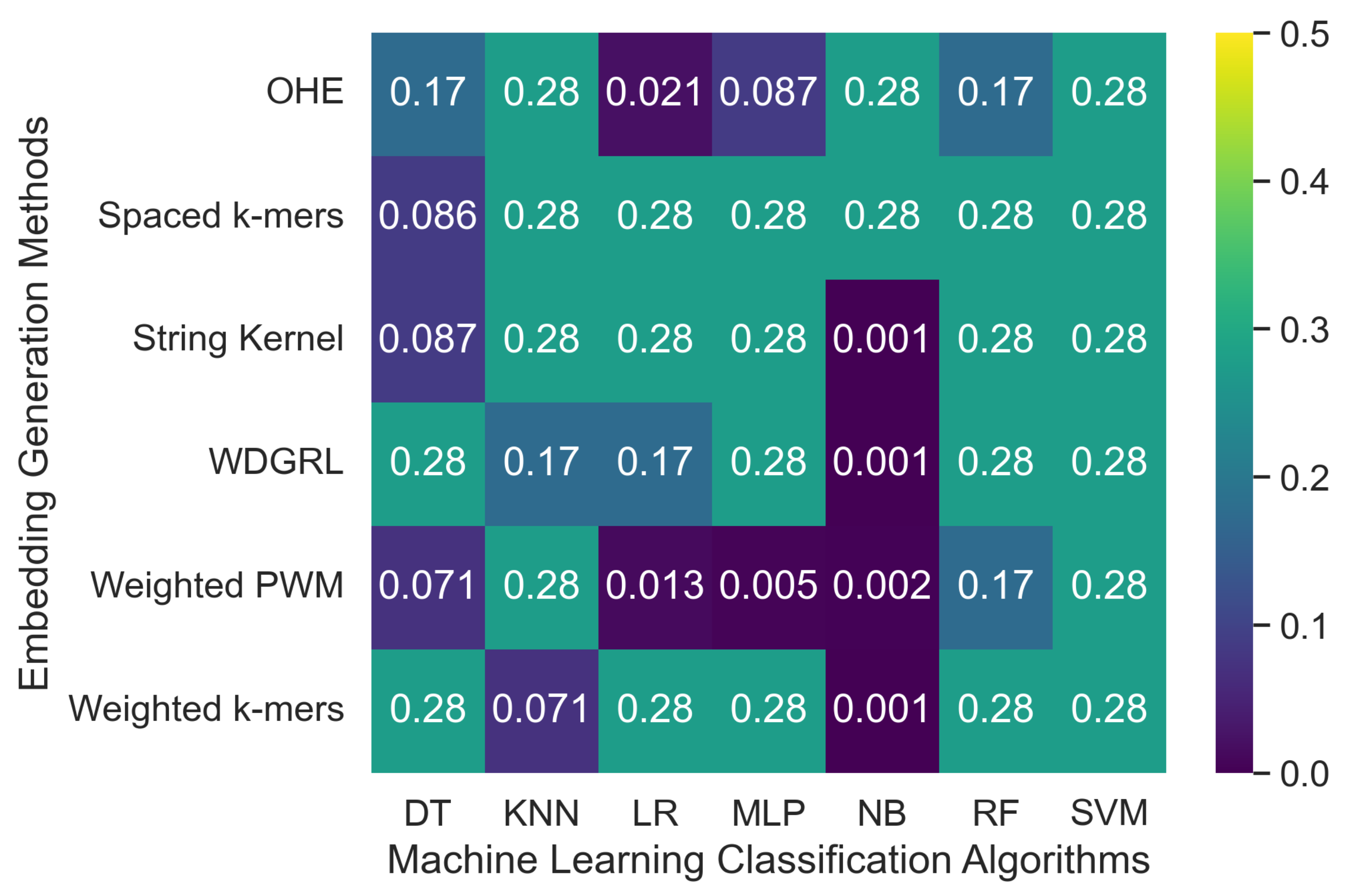
3.2.1. The Robustness Results for PacBio Sequencing Error-Incorporated Datasets
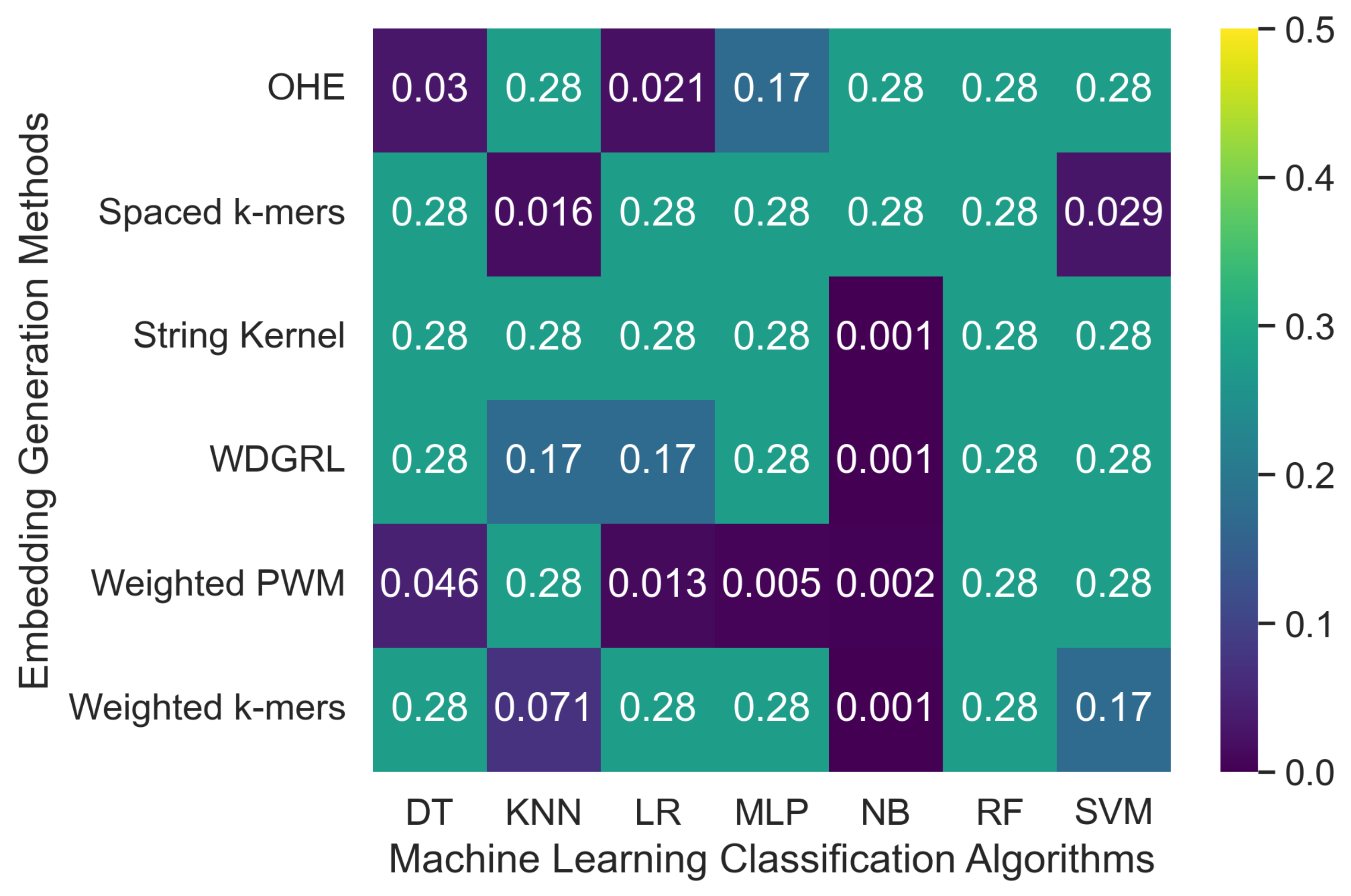
3.2.2. The Robustness Results for Oxford Nanopore Technologies (ONT) Sequencing Error-Incorporated Datasets
3.2.3. The Robustness Results for Random-Error-Incorporated Datasets
3.3. Comparison of Predictive Performance of Machine Learning Models on SARS-CoV-2 Sequences with Errors from PacBio and ONT Sequencers
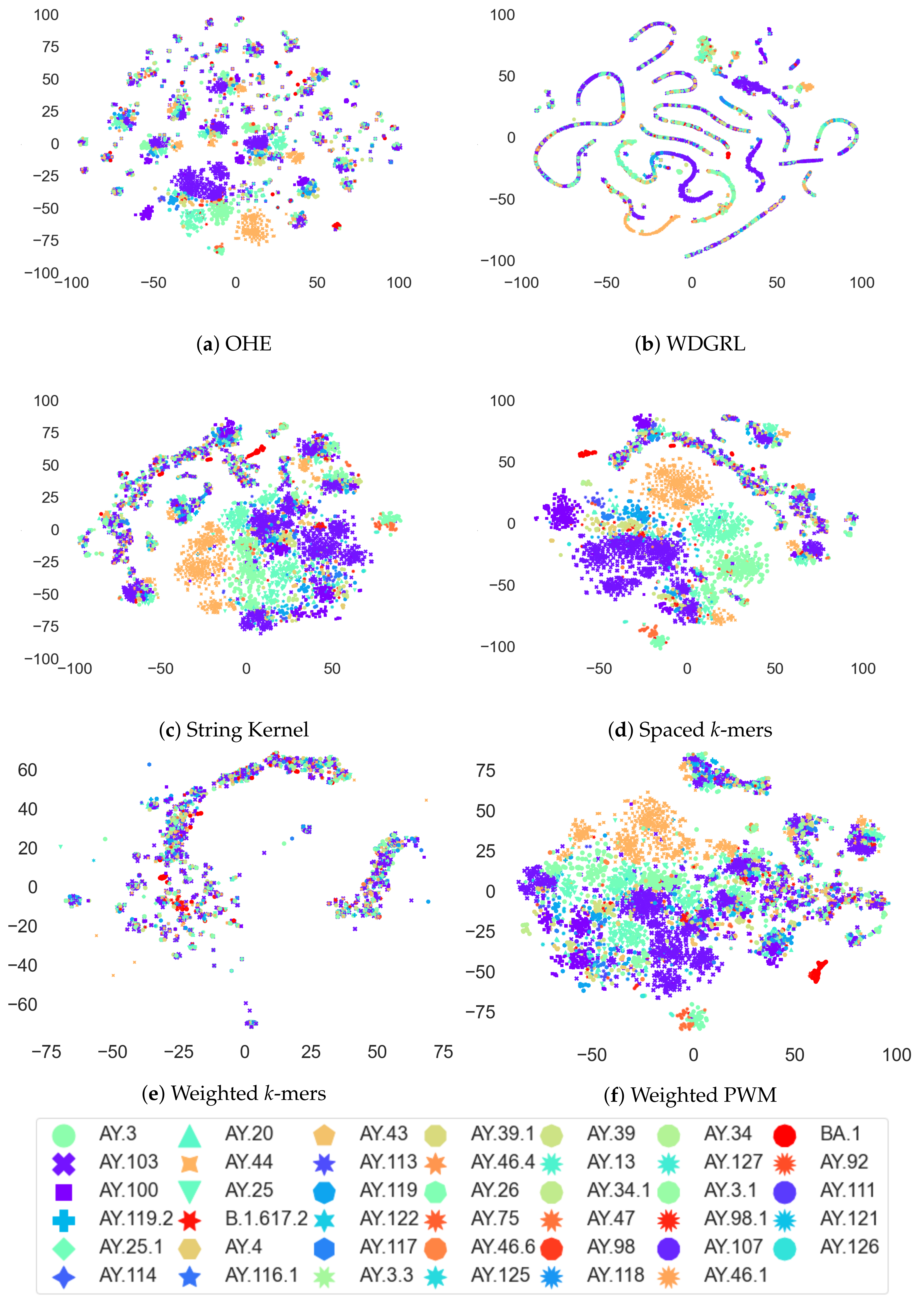
3.4. Analysis of Coronavirus Variants Based on Different Embedding Vector Generation Methods Using t-SNE Visualization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| WGS | Whole Genome Sequencing |
| TGS | Third-Generation Sequencing |
| GISAID | Global Initiative on Sharing All Influenza Data |
| ML | Machine Learning |
| DL | Deep Learning |
| WDGRL | Wasserstein Distance-based Generative Adversarial Network for Representation Learning |
| PacBio | Pacific Biosciences |
| ONT | Oxford Nanopore Technologies |
| OHE | One-Hot Encoding |
| PWM | Position Weight Matrix |
| SVM | Support Vector Machine |
| NB | Naïve Bayes |
| MLP | Multi-Layer Perceptron |
| KNN | K-Nearest Neighbors |
| RF | Random Forest |
| LR | Logistic Regression |
| DT | Decision Tree |
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E. Epidemiology, virology, and clinical features of severe acute respiratory syndrome -coronavirus-2 (SARS-CoV-2; Coronavirus Disease-19). Clin. Exp. Pediatr. 2020, 63, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- GISAID Website. 2021. Available online: https://www.gisaid.org/ (accessed on 9 June 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Reviews. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Arons, M.M.; Hatfield, K.M.; Reddy, S.C.; Kimball, A.; James, A.; Jacobs, J.R.; Taylor, J.; Spicer, K.; Bardossy, A.C.; Oakley, L.P.; et al. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N. Engl. J. Med. 2020, 382, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.; Gnanakaran, S. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laver, T.; Harrison, J.; O’Neill, P.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, A. Third Generation DNA Sequencing: Pacific Biosciences’ Single Molecule Real Time Technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, J.; Spacek, D.V.; Snyder, M. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, O.P.; Vallejo, M.; El-Badawy, I.M.; Aysha, A.; Madhanagopal, J.; Mohd Faudzi, A.A. Classification of SARS-CoV-2 and non-SARS-CoV-2 using machine learning algorithms. Comput. Biol. Med. 2021, 136, 104650. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Sahoo, B.; Ullah, N.; Zelikovskiy, A.; Patterson, M.; Khan, I. A k-mer based approach for sars-cov-2 variant identification. In Proceedings of the International Symposium on Bioinformatics Research and Applications, Shenzhen, China, 26–28 November 2021; pp. 153–164. [Google Scholar]
- Kuzmin, K.; Adeniyi, A.E.; DaSouza, A.K., Jr.; Lim, D.; Nguyen, H.; Molina, N.R.; Xiong, L.; Weber, I.T.; Harrison, R.W. Machine learning methods accurately predict host specificity of coronaviruses based on spike sequences alone. Biochem. Biophys. Res. Commun. 2020, 533, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Sekhon, A.; Kowsari, K.; Lanchantin, J.; Wang, B.; Qi, Y. Gakco: A fast gapped k-mer string kernel using counting. In Proceedings of the Joint European Conference on Machine Learning and Knowledge Discovery in Databases, Skopje, Macedonia, 18–22 September 2017; pp. 356–373. [Google Scholar]
- Ali, S.; Sahoo, B.; Zelikovsky, A.; Chen, P.Y.; Patterson, M. Benchmarking machine learning robustness in COVID-19 genome sequence classification. Sci. Rep. 2023, 13, 4154. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Asai, K.; Hamada, M. PBSIM: PacBio reads simulator—toward accurate genome assembly. Bioinformatics 2012, 29, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Wick, R. Badread: Simulation of error-prone long reads. J. Open Source Softw. 2019, 4, 1316. [Google Scholar] [CrossRef]
- Shen, J.; Qu, Y.; Zhang, W.; Yu, Y. Wasserstein distance guided representation learning for domain adaptation. In Proceedings of the AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Farhan, M.; Tariq, J.; Zaman, A.; Shabbir, M.; Khan, I. Efficient Approximation Algorithms for Strings Kernel Based Sequence Classification. In Proceedings of the Advances in Neural Information Processing Systems (NeurIPS), Long Beach, CA, USA, 4–9 December 2017; pp. 6935–6945. [Google Scholar]
- Ali, S.; Bello, B.; Chourasia, P.; Punathil, R.T.; Zhou, Y.; Patterson, M. PWM2Vec: An Efficient Embedding Approach for Viral Host Specification from Coronavirus Spike Sequences. Biology 2022, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 2014, 15, 3221–3245. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | No. Sequences | Lineage | No. Sequences |
|---|---|---|---|
| AY.103 | 2253 | AY.121 | 40 |
| AY.44 | 1407 | AY.75 | 36 |
| AY.100 | 715 | AY.3.1 | 30 |
| AY.3 | 705 | AY.3.3 | 28 |
| AY.25 | 582 | AY.107 | 27 |
| AY.25.1 | 381 | AY.34.1 | 25 |
| AY.39 | 247 | AY.46.6 | 21 |
| AY.119 | 241 | AY.98.1 | 20 |
| B.1.617.2 | 173 | AY.13 | 19 |
| AY.20 | 129 | AY.116.1 | 18 |
| AY.26 | 107 | AY.126 | 17 |
| AY.4 | 99 | AY.114 | 15 |
| AY.117 | 93 | AY.46.1 | 14 |
| AY.113 | 92 | AY.34 | 14 |
| AY.118 | 86 | AY.125 | 14 |
| AY.43 | 85 | AY.92 | 13 |
| AY.122 | 84 | AY.46.4 | 12 |
| BA.1 | 79 | AY.98 | 12 |
| AY.119.2 | 74 | AY.127 | 12 |
| AY.47 | 73 | AY.111 | 10 |
| AY.39.1 | 70 | _ | _ |
| Embed. Method | ML Algo. | Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) |
|---|---|---|---|---|---|---|---|---|
| OHE | SVM | 0.773 | 0.772 | 0.773 | 0.766 | 0.571 | 0.760 | 19,231.462 |
| NB | 0.086 | 0.192 | 0.086 | 0.091 | 0.194 | 0.595 | 615.813 | |
| MLP | 0.360 | 0.344 | 0.360 | 0.252 | 0.043 | 0.514 | 1222.237 | |
| KNN | 0.669 | 0.689 | 0.669 | 0.649 | 0.409 | 0.666 | 38.431 | |
| RF | 0.774 | 0.774 | 0.774 | 0.765 | 0.574 | 0.758 | 224.910 | |
| LR | 0.721 | 0.741 | 0.721 | 0.707 | 0.555 | 0.741 | 37,539.362 | |
| DT | 0.844 | 0.845 | 0.844 | 0.842 | 0.610 | 0.796 | 219.236 | |
| WDGRL | SVM | 0.327 | 0.159 | 0.327 | 0.203 | 0.036 | 0.511 | 2.789 |
| NB | 0.166 | 0.169 | 0.166 | 0.167 | 0.018 | 0.510 | 0.028 | |
| MLP | 0.413 | 0.318 | 0.413 | 0.327 | 0.077 | 0.530 | 21.971 | |
| KNN | 0.463 | 0.431 | 0.463 | 0.437 | 0.192 | 0.581 | 0.118 | |
| RF | 0.449 | 0.446 | 0.449 | 0.445 | 0.207 | 0.601 | 1.671 | |
| LR | 0.323 | 0.261 | 0.323 | 0.201 | 0.036 | 0.510 | 0.752 | |
| DT | 0.440 | 0.441 | 0.440 | 0.438 | 0.195 | 0.596 | 0.028 | |
| String Kernel | SVM | 0.881 | 0.880 | 0.881 | 0.878 | 0.776 | 0.880 | 7.200 |
| NB | 0.033 | 0.309 | 0.033 | 0.032 | 0.048 | 0.531 | 0.556 | |
| MLP | 0.715 | 0.700 | 0.715 | 0.704 | 0.369 | 0.690 | 34.899 | |
| KNN | 0.749 | 0.757 | 0.749 | 0.735 | 0.544 | 0.738 | 0.648 | |
| RF | 0.672 | 0.750 | 0.672 | 0.634 | 0.395 | 0.652 | 10.258 | |
| LR | 0.864 | 0.858 | 0.864 | 0.854 | 0.675 | 0.817 | 50.100 | |
| DT | 0.572 | 0.578 | 0.572 | 0.573 | 0.337 | 0.669 | 3.636 | |
| Spaced k-mers | SVM | 0.956 | 0.956 | 0.956 | 0.955 | 0.890 | 0.933 | 8.761 |
| NB | 0.068 | 0.305 | 0.068 | 0.059 | 0.184 | 0.606 | 0.553 | |
| MLP | 0.825 | 0.832 | 0.825 | 0.826 | 0.539 | 0.771 | 14.855 | |
| KNN | 0.796 | 0.808 | 0.796 | 0.784 | 0.611 | 0.776 | 0.754 | |
| RF | 0.915 | 0.920 | 0.915 | 0.908 | 0.749 | 0.835 | 2.107 | |
| LR | 0.956 | 0.956 | 0.956 | 0.954 | 0.881 | 0.921 | 19.964 | |
| DT | 0.834 | 0.836 | 0.834 | 0.832 | 0.647 | 0.816 | 0.739 | |
| Weighted k-mers | SVM | 0.293 | 0.201 | 0.293 | 0.158 | 0.037 | 0.510 | 30.304 |
| NB | 0.017 | 0.014 | 0.017 | 0.014 | 0.027 | 0.523 | 0.088 | |
| MLP | 0.275 | 0.168 | 0.275 | 0.159 | 0.037 | 0.511 | 16.368 | |
| KNN | 0.193 | 0.186 | 0.193 | 0.159 | 0.052 | 0.516 | 0.661 | |
| RF | 0.278 | 0.198 | 0.278 | 0.180 | 0.061 | 0.519 | 1.066 | |
| LR | 0.285 | 0.178 | 0.285 | 0.164 | 0.043 | 0.513 | 1.648 | |
| DT | 0.265 | 0.174 | 0.265 | 0.174 | 0.055 | 0.517 | 0.064 | |
| Weighted PWM | SVM | 0.852 | 0.850 | 0.852 | 0.849 | 0.741 | 0.863 | 3.257 |
| NB | 0.028 | 0.015 | 0.028 | 0.018 | 0.048 | 0.553 | 0.093 | |
| MLP | 0.760 | 0.740 | 0.760 | 0.748 | 0.393 | 0.695 | 28.944 | |
| KNN | 0.751 | 0.755 | 0.751 | 0.738 | 0.555 | 0.743 | 0.635 | |
| RF | 0.801 | 0.830 | 0.801 | 0.783 | 0.622 | 0.759 | 3.285 | |
| LR | 0.861 | 0.859 | 0.861 | 0.856 | 0.755 | 0.882 | 8.702 | |
| DT | 0.651 | 0.662 | 0.651 | 0.653 | 0.391 | 0.706 | 0.450 |
| Embed. Method | ML Algo. | Depth-5 Simulated Error | Depth-10 Simulated Error | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) | Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC- AUC | Train. Runtime (s) | ||
| OHE | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 181,443.9 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 188,143.3 |
| NB | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1452.37 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1450.059 | |
| MLP | 0.087 | 0.008 | 0.087 | 0.014 | 0.004 | 0.500 | 2833.82 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1763.060 | |
| KNN | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 126.632 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 119.041 | |
| RF | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 300.639 | 0.276 | 0.220 | 0.276 | 0.120 | 0.011 | 0.500 | 306.980 | |
| LR | 0.021 | 0.000 | 0.021 | 0.001 | 0.001 | 0.500 | 77099.6 | 0.021 | 0.000 | 0.021 | 0.001 | 0.001 | 0.500 | 71666.1 | |
| DT | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 890.873 | 0.030 | 0.087 | 0.030 | 0.002 | 0.001 | 0.500 | 441.523 | |
| WDGRL | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 8.056 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 8.117 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.053 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.052 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 9.374 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 16.305 | |
| KNN | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1.724 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1.936 | |
| RF | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.711 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.552 | |
| LR | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 0.500 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 0.574 | |
| DT | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.006 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.006 | |
| String Kernel | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 19.994 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 18.068 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 1.873 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 1.957 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 71.414 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 59.946 | |
| KNN | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.390 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.409 | |
| RF | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 14.867 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 16.127 | |
| LR | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 75.829 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 75.556 | |
| DT | 0.087 | 0.008 | 0.087 | 0.014 | 0.004 | 0.500 | 6.013 | 0.029 | 0.001 | 0.029 | 0.002 | 0.001 | 0.500 | 5.979 | |
| Spaced k-mers | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 73.149 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 60.317 |
| NB | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 6.302 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 4.487 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 126.596 | 0.016 | 0.000 | 0.016 | 0.000 | 0.001 | 0.500 | 101.059 | |
| KNN | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1.945 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.076 | |
| RF | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 8.278 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 3.487 | |
| LR | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 23.153 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 19.397 | |
| DT | 0.086 | 0.007 | 0.086 | 0.014 | 0.004 | 0.500 | 1.455 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 0.433 | |
| Weighted k-mers | SVM | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 63.260 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 62.970 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.870 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.697 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 29.469 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 25.876 | |
| KNN | 0.071 | 0.005 | 0.071 | 0.009 | 0.003 | 0.500 | 1.970 | 0.071 | 0.005 | 0.071 | 0.009 | 0.003 | 0.500 | 1.979 | |
| RF | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 1.683 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 1.640 | |
| LR | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 2.404 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 2.378 | |
| DT | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 0.098 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 0.097 | |
| Weighted PWM | SVM | 0.276 | 0.079 | 0.276 | 0.120 | 0.013 | 0.501 | 8.651 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 10.205 |
| NB | 0.002 | 0.000 | 0.002 | 0.000 | 0.000 | 0.500 | 0.787 | 0.002 | 0.000 | 0.002 | 0.000 | 0.000 | 0.500 | 0.622 | |
| MLP | 0.005 | 0.000 | 0.005 | 0.000 | 0.000 | 0.500 | 24.250 | 0.005 | 0.000 | 0.005 | 0.000 | 0.000 | 0.500 | 31.357 | |
| KNN | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 2.358 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 2.828 | |
| RF | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 4.826 | 0.276 | 0.076 | 0.276 | 0.120 | 0.011 | 0.500 | 6.342 | |
| LR | 0.013 | 0.000 | 0.013 | 0.000 | 0.001 | 0.500 | 13.294 | 0.013 | 0.000 | 0.013 | 0.000 | 0.001 | 0.500 | 17.801 | |
| DT | 0.071 | 0.005 | 0.071 | 0.009 | 0.003 | 0.500 | 0.694 | 0.046 | 0.002 | 0.046 | 0.004 | 0.002 | 0.500 | 0.793 | |
| Embed. Method | ML Algo. | n2020 5× Simulated Error | n2020 10× Simulated Error | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) | Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) | ||
| OHE | SVM | 0.122 | 0.133 | 0.122 | 0.112 | 0.022 | 0.501 | 70,679.0 | 0.106 | 0.127 | 0.106 | 0.094 | 0.018 | 0.500 | 103,909.2 |
| NB | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1075.20 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 833.594 | |
| MLP | 0.071 | 0.097 | 0.071 | 0.010 | 0.003 | 0.500 | 3906.45 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1539.707 | |
| KNN | 0.189 | 0.130 | 0.189 | 0.134 | 0.019 | 0.501 | 82.195 | 0.189 | 0.133 | 0.189 | 0.134 | 0.017 | 0.500 | 108.649 | |
| RF | 0.072 | 0.141 | 0.072 | 0.076 | 0.008 | 0.500 | 276.773 | 0.241 | 0.116 | 0.241 | 0.147 | 0.016 | 0.500 | 319.020 | |
| LR | 0.272 | 0.108 | 0.272 | 0.122 | 0.011 | 0.500 | 67103.9 | 0.270 | 0.130 | 0.270 | 0.124 | 0.012 | 0.500 | 68286.4 | |
| DT | 0.166 | 0.134 | 0.166 | 0.103 | 0.017 | 0.501 | 460.552 | 0.163 | 0.127 | 0.163 | 0.087 | 0.013 | 0.500 | 411.342 | |
| WDGRL | SVM | 0.244 | 0.108 | 0.244 | 0.147 | 0.015 | 0.501 | 8.906 | 0.214 | 0.119 | 0.214 | 0.138 | 0.016 | 0.502 | 8.812 |
| NB | 0.011 | 0.094 | 0.011 | 0.019 | 0.002 | 0.496 | 0.063 | 0.184 | 0.129 | 0.184 | 0.089 | 0.011 | 0.501 | 0.060 | |
| MLP | 0.268 | 0.085 | 0.268 | 0.122 | 0.012 | 0.500 | 30.652 | 0.192 | 0.137 | 0.192 | 0.116 | 0.013 | 0.502 | 28.833 | |
| KNN | 0.261 | 0.150 | 0.261 | 0.127 | 0.013 | 0.500 | 0.382 | 0.185 | 0.145 | 0.185 | 0.120 | 0.018 | 0.502 | 0.374 | |
| RF | 0.109 | 0.147 | 0.109 | 0.080 | 0.012 | 0.499 | 2.572 | 0.153 | 0.151 | 0.153 | 0.090 | 0.014 | 0.500 | 2.383 | |
| LR | 0.125 | 0.073 | 0.125 | 0.074 | 0.009 | 0.500 | 1.088 | 0.191 | 0.167 | 0.191 | 0.088 | 0.011 | 0.501 | 1.058 | |
| DT | 0.103 | 0.148 | 0.103 | 0.074 | 0.012 | 0.499 | 0.048 | 0.166 | 0.157 | 0.166 | 0.093 | 0.014 | 0.501 | 0.047 | |
| String Kernel | SVM | 0.144 | 0.134 | 0.144 | 0.137 | 0.024 | 0.500 | 20.179 | 0.154 | 0.142 | 0.154 | 0.147 | 0.025 | 0.501 | 18.168 |
| NB | 0.004 | 0.000 | 0.004 | 0.000 | 0.001 | 0.503 | 2.019 | 0.004 | 0.000 | 0.004 | 0.000 | 0.001 | 0.499 | 1.803 | |
| MLP | 0.137 | 0.135 | 0.137 | 0.135 | 0.026 | 0.501 | 59.144 | 0.132 | 0.139 | 0.132 | 0.134 | 0.028 | 0.502 | 49.297 | |
| KNN | 0.195 | 0.123 | 0.195 | 0.132 | 0.020 | 0.500 | 2.877 | 0.189 | 0.137 | 0.189 | 0.147 | 0.022 | 0.500 | 2.807 | |
| RF | 0.249 | 0.116 | 0.249 | 0.144 | 0.015 | 0.500 | 15.951 | 0.263 | 0.157 | 0.263 | 0.153 | 0.016 | 0.501 | 15.812 | |
| LR | 0.157 | 0.130 | 0.157 | 0.141 | 0.023 | 0.500 | 78.382 | 0.171 | 0.140 | 0.171 | 0.153 | 0.024 | 0.500 | 76.744 | |
| DT | 0.130 | 0.129 | 0.130 | 0.129 | 0.023 | 0.500 | 5.910 | 0.137 | 0.140 | 0.137 | 0.137 | 0.026 | 0.501 | 5.969 | |
| Spaced k-mers | SVM | 0.132 | 0.147 | 0.132 | 0.133 | 0.039 | 0.508 | 23.890 | 0.193 | 0.208 | 0.193 | 0.198 | 0.092 | 0.535 | 20.470 |
| NB | 0.004 | 0.000 | 0.004 | 0.000 | 0.002 | 0.503 | 3.843 | 0.006 | 0.003 | 0.006 | 0.001 | 0.004 | 0.502 | 4.476 | |
| MLP | 0.112 | 0.142 | 0.112 | 0.118 | 0.025 | 0.502 | 58.597 | 0.176 | 0.192 | 0.176 | 0.178 | 0.051 | 0.514 | 44.872 | |
| KNN | 0.225 | 0.150 | 0.225 | 0.146 | 0.018 | 0.500 | 3.024 | 0.222 | 0.177 | 0.222 | 0.181 | 0.035 | 0.508 | 2.920 | |
| RF | 0.206 | 0.148 | 0.206 | 0.162 | 0.033 | 0.506 | 4.026 | 0.232 | 0.196 | 0.232 | 0.207 | 0.062 | 0.519 | 4.038 | |
| LR | 0.135 | 0.151 | 0.135 | 0.137 | 0.040 | 0.509 | 70.516 | 0.198 | 0.211 | 0.198 | 0.202 | 0.097 | 0.538 | 57.721 | |
| DT | 0.131 | 0.140 | 0.131 | 0.128 | 0.028 | 0.502 | 1.050 | 0.145 | 0.176 | 0.145 | 0.157 | 0.050 | 0.516 | 1.011 | |
| Weighted k-mers | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 73.858 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 74.108 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.778 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.805 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 30.145 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 31.810 | |
| KNN | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 2.079 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1.937 | |
| RF | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1.763 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1.645 | |
| LR | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.557 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.473 | |
| DT | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.116 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.116 | |
| Weighted PWM | SVM | 0.203 | 0.141 | 0.203 | 0.157 | 0.023 | 0.501 | 11.123 | 0.240 | 0.170 | 0.240 | 0.177 | 0.028 | 0.504 | 9.825 |
| NB | 0.002 | 0.000 | 0.002 | 0.000 | 0.000 | 0.498 | 0.808 | 0.003 | 0.000 | 0.003 | 0.000 | 0.000 | 0.500 | 0.814 | |
| MLP | 0.086 | 0.141 | 0.086 | 0.091 | 0.022 | 0.501 | 36.325 | 0.119 | 0.180 | 0.119 | 0.132 | 0.035 | 0.513 | 27.304 | |
| KNN | 0.193 | 0.127 | 0.193 | 0.129 | 0.017 | 0.501 | 2.745 | 0.188 | 0.148 | 0.188 | 0.151 | 0.025 | 0.503 | 2.475 | |
| RF | 0.175 | 0.141 | 0.175 | 0.151 | 0.031 | 0.505 | 5.887 | 0.203 | 0.175 | 0.203 | 0.181 | 0.042 | 0.510 | 4.699 | |
| LR | 0.084 | 0.152 | 0.084 | 0.093 | 0.026 | 0.505 | 16.420 | 0.143 | 0.194 | 0.143 | 0.155 | 0.051 | 0.519 | 12.654 | |
| DT | 0.069 | 0.128 | 0.069 | 0.077 | 0.021 | 0.501 | 0.830 | 0.101 | 0.152 | 0.101 | 0.115 | 0.030 | 0.504 | 0.670 | |
| Embed. Method | ML Algo. | Random 5× Simulated Error | Random 10× Simulated Error | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) | Acc. | Prec. | Recall | F1 Weigh. | F1 Macro | ROC-AUC | Train. Runtime (s) | ||
| OHE | SVM | 0.101 | 0.138 | 0.101 | 0.078 | 0.016 | 0.500 | 157054.2 | 0.107 | 0.145 | 0.107 | 0.093 | 0.019 | 0.501 | 74,697.7 |
| NB | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1113.271 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1119.210 | |
| MLP | 0.013 | 0.000 | 0.013 | 0.000 | 0.002 | 0.500 | 1503.268 | 0.267 | 0.085 | 0.267 | 0.125 | 0.012 | 0.500 | 1625.962 | |
| KNN | 0.223 | 0.131 | 0.223 | 0.136 | 0.018 | 0.500 | 91.708 | 0.204 | 0.135 | 0.204 | 0.139 | 0.018 | 0.501 | 100.443 | |
| RF | 0.202 | 0.112 | 0.202 | 0.126 | 0.014 | 0.500 | 259.562 | 0.186 | 0.114 | 0.186 | 0.112 | 0.013 | 0.500 | 405.555 | |
| LR | 0.271 | 0.109 | 0.271 | 0.122 | 0.011 | 0.500 | 70035.07 | 0.272 | 0.123 | 0.272 | 0.122 | 0.012 | 0.500 | 83434.3 | |
| DT | 0.157 | 0.126 | 0.157 | 0.075 | 0.012 | 0.500 | 776.263 | 0.164 | 0.136 | 0.164 | 0.086 | 0.014 | 0.500 | 943.475 | |
| WDGRL | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 8.776 | 0.275 | 0.118 | 0.275 | 0.158 | 0.017 | 0.502 | 8.842 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.068 | 0.005 | 0.105 | 0.005 | 0.007 | 0.001 | 0.500 | 0.051 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 31.487 | 0.272 | 0.129 | 0.272 | 0.125 | 0.012 | 0.500 | 21.765 | |
| KNN | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.348 | 0.169 | 0.142 | 0.169 | 0.119 | 0.016 | 0.500 | 0.383 | |
| RF | 0.087 | 0.008 | 0.087 | 0.014 | 0.004 | 0.500 | 2.488 | 0.072 | 0.138 | 0.072 | 0.086 | 0.015 | 0.499 | 2.510 | |
| LR | 0.087 | 0.008 | 0.087 | 0.014 | 0.004 | 0.500 | 1.069 | 0.267 | 0.115 | 0.267 | 0.127 | 0.012 | 0.500 | 1.059 | |
| DT | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.046 | 0.068 | 0.135 | 0.068 | 0.083 | 0.015 | 0.499 | 0.046 | |
| String Kernel | SVM | 0.169 | 0.132 | 0.169 | 0.142 | 0.024 | 0.501 | 19.308 | 0.140 | 0.128 | 0.140 | 0.133 | 0.023 | 0.500 | 18.115 |
| NB | 0.003 | 0.004 | 0.003 | 0.001 | 0.002 | 0.501 | 1.894 | 0.004 | 0.000 | 0.004 | 0.000 | 0.001 | 0.500 | 1.811 | |
| MLP | 0.138 | 0.135 | 0.138 | 0.134 | 0.023 | 0.500 | 83.035 | 0.124 | 0.130 | 0.124 | 0.127 | 0.024 | 0.500 | 60.808 | |
| KNN | 0.213 | 0.128 | 0.213 | 0.130 | 0.017 | 0.500 | 2.773 | 0.211 | 0.136 | 0.211 | 0.142 | 0.020 | 0.501 | 2.816 | |
| RF | 0.263 | 0.140 | 0.263 | 0.140 | 0.014 | 0.500 | 15.649 | 0.257 | 0.122 | 0.257 | 0.146 | 0.015 | 0.500 | 15.715 | |
| LR | 0.180 | 0.132 | 0.180 | 0.148 | 0.022 | 0.500 | 79.243 | 0.158 | 0.129 | 0.158 | 0.141 | 0.022 | 0.500 | 76.412 | |
| DT | 0.134 | 0.129 | 0.134 | 0.129 | 0.023 | 0.500 | 6.318 | 0.125 | 0.130 | 0.125 | 0.127 | 0.022 | 0.499 | 5.773 | |
| Spaced k-mers | SVM | 0.080 | 0.139 | 0.080 | 0.080 | 0.012 | 0.501 | 17.010 | 0.124 | 0.155 | 0.124 | 0.118 | 0.022 | 0.501 | 22.282 |
| NB | 0.005 | 0.004 | 0.005 | 0.001 | 0.002 | 0.502 | 3.879 | 0.006 | 0.000 | 0.006 | 0.001 | 0.002 | 0.500 | 4.803 | |
| MLP | 0.133 | 0.111 | 0.133 | 0.060 | 0.011 | 0.500 | 41.950 | 0.116 | 0.163 | 0.116 | 0.107 | 0.019 | 0.502 | 47.611 | |
| KNN | 0.263 | 0.092 | 0.263 | 0.121 | 0.012 | 0.500 | 2.763 | 0.246 | 0.143 | 0.246 | 0.125 | 0.014 | 0.500 | 2.646 | |
| RF | 0.210 | 0.128 | 0.210 | 0.148 | 0.019 | 0.501 | 3.921 | 0.233 | 0.139 | 0.233 | 0.159 | 0.023 | 0.502 | 3.587 | |
| LR | 0.147 | 0.144 | 0.147 | 0.116 | 0.018 | 0.501 | 64.852 | 0.135 | 0.171 | 0.135 | 0.128 | 0.027 | 0.505 | 60.750 | |
| DT | 0.156 | 0.128 | 0.156 | 0.088 | 0.015 | 0.501 | 0.952 | 0.127 | 0.135 | 0.127 | 0.096 | 0.020 | 0.501 | 1.006 | |
| Weighted k-mers | SVM | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 76.220 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 78.497 |
| NB | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.774 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.500 | 0.749 | |
| MLP | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 23.639 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 34.219 | |
| KNN | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1.932 | 0.172 | 0.030 | 0.172 | 0.051 | 0.007 | 0.500 | 1.915 | |
| RF | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1.851 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 1.749 | |
| LR | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.706 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 2.626 | |
| DT | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.100 | 0.276 | 0.076 | 0.276 | 0.119 | 0.011 | 0.500 | 0.102 | |
| Weighted PWM | SVM | 0.203 | 0.133 | 0.203 | 0.145 | 0.020 | 0.500 | 11.330 | 0.196 | 0.143 | 0.196 | 0.151 | 0.024 | 0.501 | 8.605 |
| NB | 0.002 | 0.000 | 0.002 | 0.000 | 0.000 | 0.500 | 0.594 | 0.002 | 0.000 | 0.002 | 0.000 | 0.000 | 0.500 | 0.766 | |
| MLP | 0.016 | 0.087 | 0.016 | 0.015 | 0.004 | 0.499 | 43.680 | 0.037 | 0.110 | 0.037 | 0.036 | 0.012 | 0.501 | 29.312 | |
| KNN | 0.214 | 0.108 | 0.214 | 0.122 | 0.015 | 0.500 | 2.758 | 0.201 | 0.143 | 0.201 | 0.130 | 0.017 | 0.500 | 2.618 | |
| RF | 0.163 | 0.122 | 0.163 | 0.129 | 0.017 | 0.501 | 5.649 | 0.181 | 0.134 | 0.181 | 0.136 | 0.019 | 0.501 | 4.915 | |
| LR | 0.061 | 0.104 | 0.061 | 0.058 | 0.010 | 0.499 | 16.697 | 0.070 | 0.131 | 0.070 | 0.074 | 0.014 | 0.502 | 12.835 | |
| DT | 0.061 | 0.138 | 0.061 | 0.058 | 0.015 | 0.500 | 0.875 | 0.073 | 0.136 | 0.073 | 0.070 | 0.018 | 0.500 | 0.673 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahoo, B.; Ali, S.; Chen, P.-Y.; Patterson, M.; Zelikovsky, A. Assessing the Resilience of Machine Learning Classification Algorithms on SARS-CoV-2 Genome Sequences Generated with Long-Read Specific Errors. Biomolecules 2023, 13, 934. https://doi.org/10.3390/biom13060934
Sahoo B, Ali S, Chen P-Y, Patterson M, Zelikovsky A. Assessing the Resilience of Machine Learning Classification Algorithms on SARS-CoV-2 Genome Sequences Generated with Long-Read Specific Errors. Biomolecules. 2023; 13(6):934. https://doi.org/10.3390/biom13060934
Chicago/Turabian StyleSahoo, Bikram, Sarwan Ali, Pin-Yu Chen, Murray Patterson, and Alexander Zelikovsky. 2023. "Assessing the Resilience of Machine Learning Classification Algorithms on SARS-CoV-2 Genome Sequences Generated with Long-Read Specific Errors" Biomolecules 13, no. 6: 934. https://doi.org/10.3390/biom13060934
APA StyleSahoo, B., Ali, S., Chen, P. -Y., Patterson, M., & Zelikovsky, A. (2023). Assessing the Resilience of Machine Learning Classification Algorithms on SARS-CoV-2 Genome Sequences Generated with Long-Read Specific Errors. Biomolecules, 13(6), 934. https://doi.org/10.3390/biom13060934