
(Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters


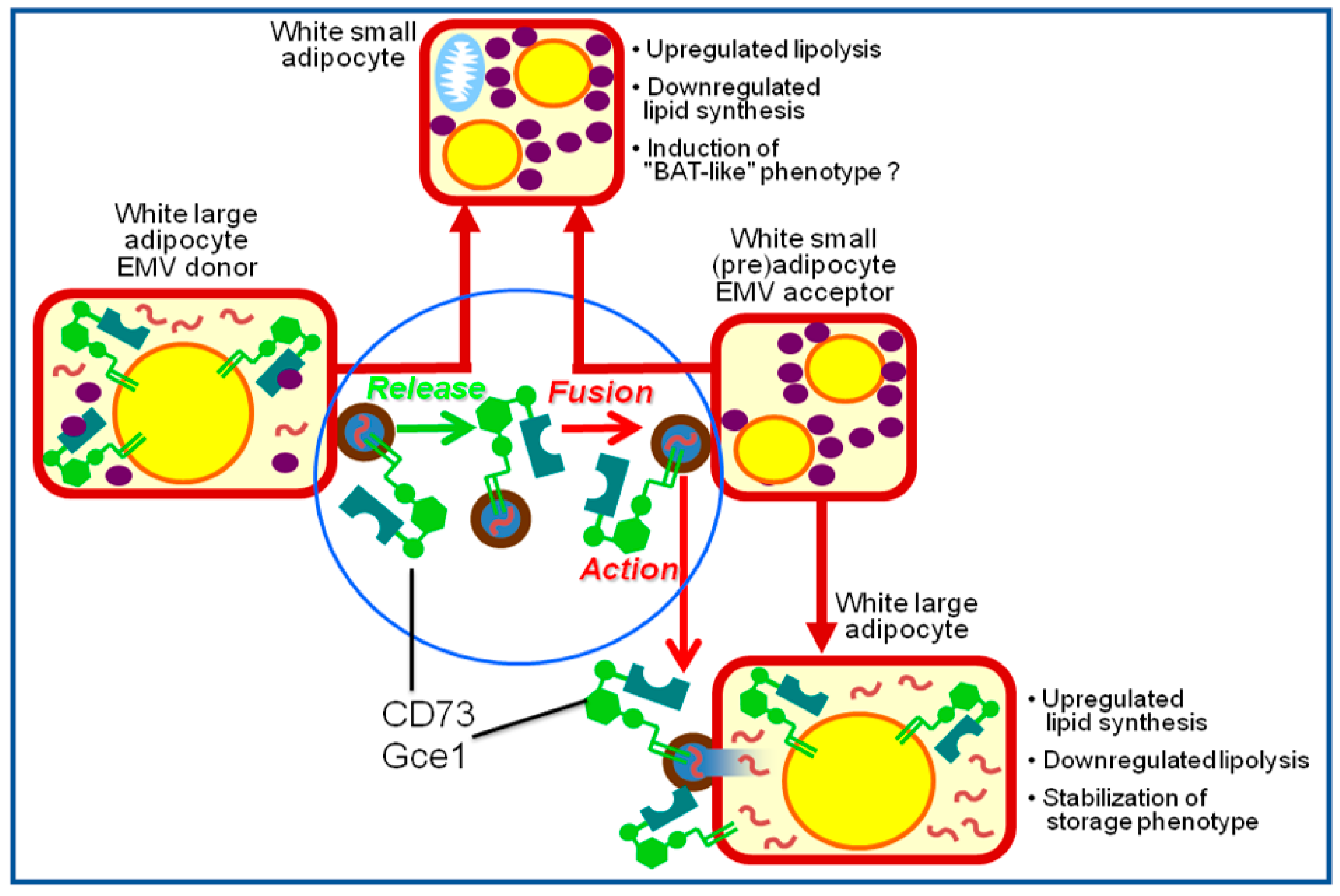{kind=link}
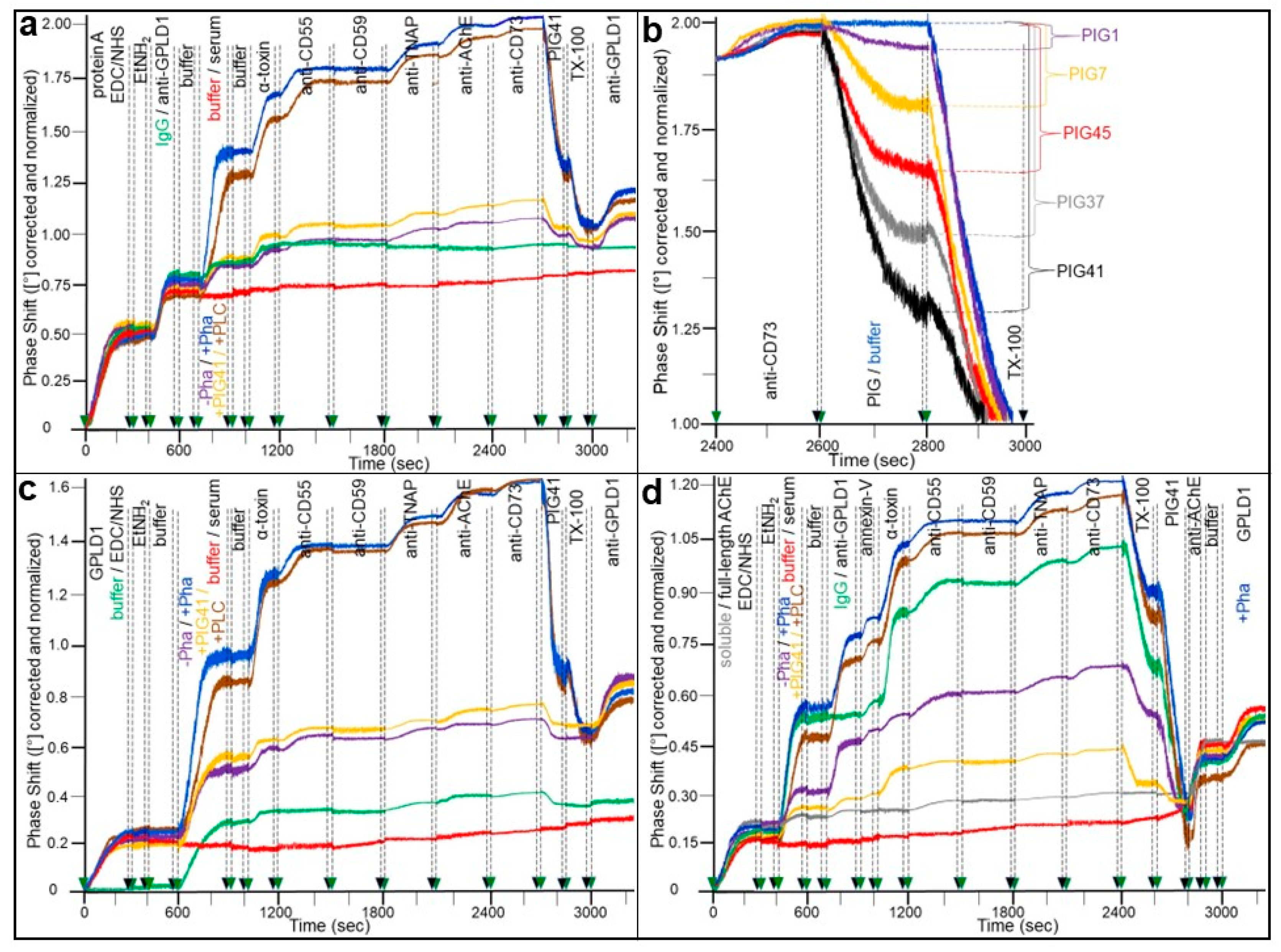{kind=link}
{kind=link}
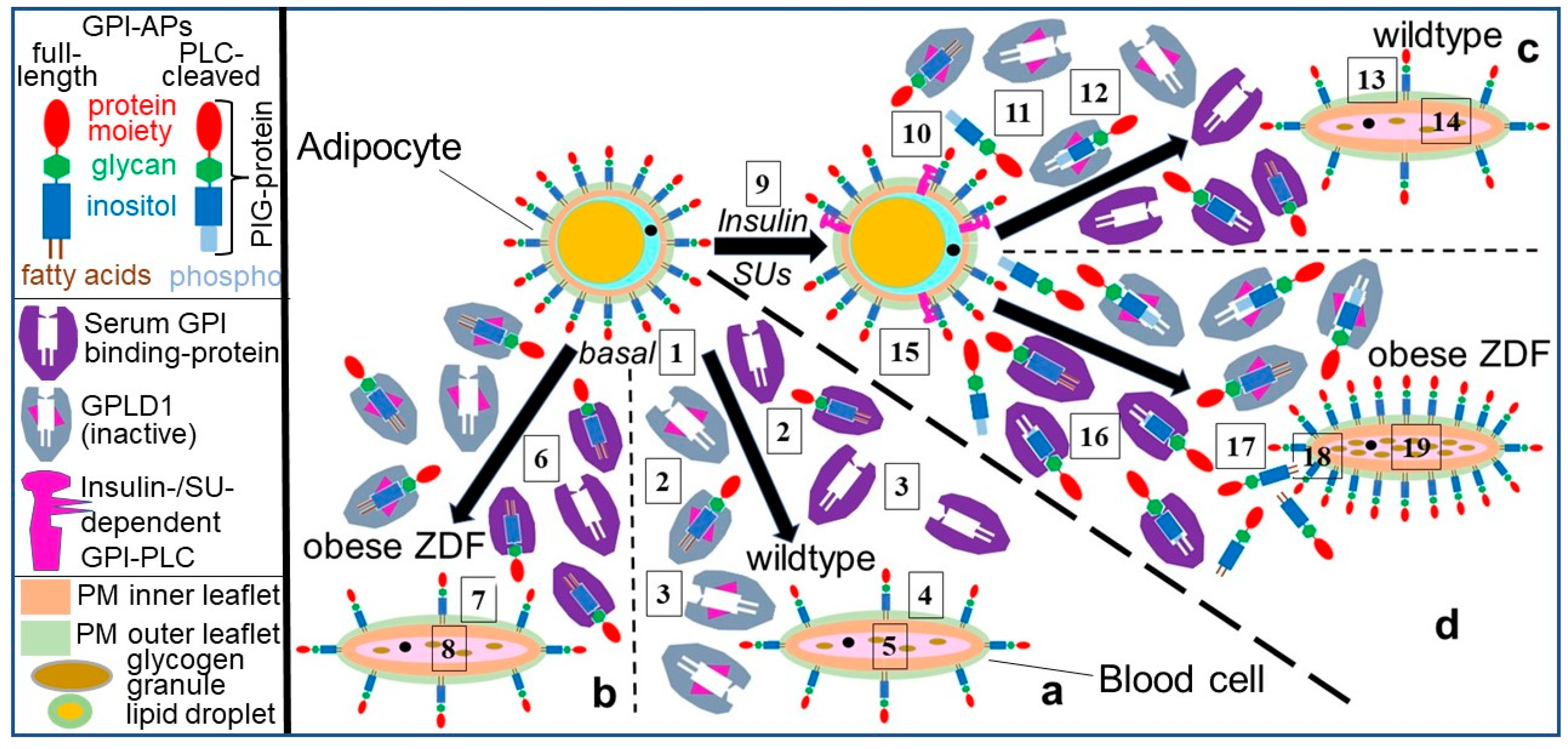{kind=link}
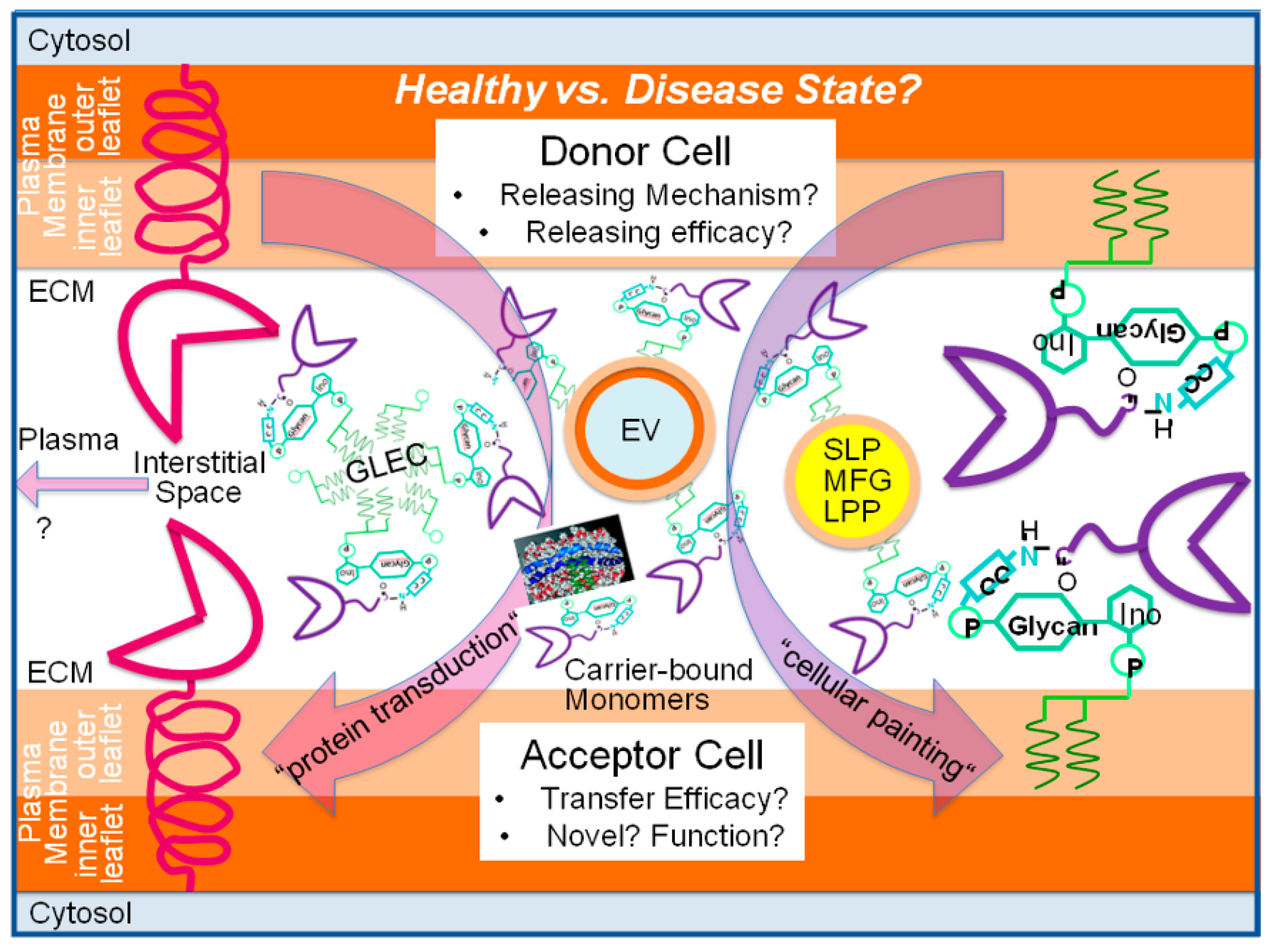{kind=link}
Abstract
:1. Introduction
2. Transfer via Vesicular Mechanisms
3. Transfer via Non-Vesicular Mechanisms
4. Transfer via Micelle-like Complexes
5. Some Thoughts about the Evolution of GPI-AP Transfer
6. Some Thoughts about PIGs, Mediators of Insulin Action, and GPI-AP Transfer
7. Transfer of Matter rather than Information
8. Non-Genetic Inheritance of Acquired Features
- (i)
- The pathogenic—donor—bacteria co-cultured with the non-pathogenic—acceptor—bacteria release certain membrane proteins (rather than DNS fragments encoding them), which confer the pathogenic surface characteristics from the PMs into the culture medium via vesicular or non-vesicular vehicles and mechanisms (rather than cell lysis).
- (ii)
- Those pathogenic membrane proteins (rather than DNS fragments) become transferred to the acceptor bacteria, which lack them.
- (iii)
- Upon incorporation of the transferred pathogenic membrane proteins into specific pre-existing “membrane landscapes” of the PMs, which operate as a matrix or template, and their correct and functional incorporation and assembly into those structures (analogous to the one DNA strand of the double helix acting as a template for the newly synthesized one), the acceptor bacteria will display the typical pathogenic phenotype as exerted by the donor bacteria.
- (iv)
- The pathogenic phenotype will be transmitted from the bacteria with acquired pathogenicity as new donors to the next non-pathogenic acceptor cells in a reliable, stable, and recurring fashion.
9. Agential Realism: A Framework for Knowledge Production for (Non-Genetic) Inheritance
- -
- the transfer of genetic and the transfer of non-genetic materials,
- -
- mother and daughter cells as well as soma and germline cells which do not rely on genes,
- -
- the inheritance of features of “historical” origin, i.e., those caused by recombination/mutation prior to the start of the life of the organism and the inheritance of features acquired during the individual life span of the organism, i.e., features resulting from—intentional or non-intentional—adaptation,
- -
- the socio-political inheritance of private property and the biological inheritance at the various stages of legislation and scientific discovery during the ages.
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eisenhaber, B.; Bork, P.; Eisenhaber, B. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: Analysis of protein sequence data from complete genomes. Protein Eng. 2001, 14, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Poisson, G.; Chauve, C.; Chen, X.; Bergeron, A. FragAnchor: A large-scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genom. Proteom. Bioinform. 2007, 5, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020, 10, 190290. [Google Scholar] [CrossRef] [Green Version]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI proteins: Sorting, membrane residence and functions. Biochim. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, Y.; Ikawa, M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016, 57, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G.A.; Müller, T.D. (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments. Biomolecules 2023, 13, 855. [Google Scholar] [CrossRef]
- Greening, D.W.; Kapp, E.A.; Ji, H.; Speed, T.P.; Simpson, R.J. Colon tumour secretopeptidome: Insights into endogenous proteolytic cleavage events in the colon tumour microenvironment. Biochim. Biophys. Acta 2013, 1834, 2396–2407. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Coomaraswamy, J.; Watts, J.C.; Yang, J.; Ko, K.W.S.; Strome, B.; Mercer, R.C.C.; Wohlgemuth, S.L.; Schmitt-Ulms, G.; Westaway, D. Endoproteolytic processing of the mammalian prion glycoprotein family. FEBS Lett. 2014, 281, 862–876. [Google Scholar] [CrossRef]
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. 2009, 14, 2494–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallone, D.; Malagolini, N.; Serafini-Cessi, F. Mechanism of release of urinary Tamm-Horsfall glycoprotein from the kidney GPI-anchored counterpart. Biochim. Biophys. Res. Commun. 2001, 280, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q. 150 years of Darwin’s theory of intercellular flow of hereditary information. Nature Rev. Mol. Cell Biol. 2018, 19, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Darwin, C.R. Pangenesis: Mr. Darwin’s reply to Professor Delpino scientific opinion. Sci. Opin. 1869, 2, 426. [Google Scholar]
- Darwin, C. Pangenesis. Nature 1871, 3, 502. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Mathew, A.; Mason, A.B.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalisation of obsolete membrane proteins. J. Cell. Pathol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Johnstone, R.M. Revisiting the road to the discovery of exosomes. Blood Cells Mol. Dis. 2003, 34, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell. Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.Y.; Naora, H. Extracellular vesicle membrane-associated proteins: Emerging roles in tumor angiogenesis and anti-angiogenesis therapy resistance. Int. J. Mol. Sci. 2020, 21, 5418. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Gong, H.; Luo, S.; Cui, Y. The role of exosomes and their applications in cancer. Int. J. Mol. Sci. 2021, 22, 12204. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a new frontier of cancer liquid biopsy. Mol. Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Müller, G.A. Glycosylphosphatidylinositol-Anchored Proteins and Their Release from Cells—From Phenomenon to Meaning, 1st ed.; Nova Science Publishers—Biochemistry Research Trends: New York, NY, USA, 2018; pp. 104–115. [Google Scholar]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Isaac, R.; Reis, F.C.G.; Ying, W.; Olefsky, J. Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab. 2021, 33, 1744–1762. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.N.; Chen, J.; Zhang, C.J.; Xie, X.J.; Liao, D.F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell. Commun. Signal. 2020, 18, 119. [Google Scholar] [CrossRef]
- Kirchhoff, C.; Hale, G. Cell-to-cell transfer of glycosylphosphatidylinositol-anchored membrane proteins during sperm maturation. Mol. Hum. Reprod. 1996, 2, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Leßleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaaau6977. [Google Scholar] [CrossRef] [PubMed]
- Pap, E.; Pallinger, E.; Pasztoi, M.; Falus, A. Highlights of a new type of intercellular communication: Microvesicle-based information transfer. Inflamm. Res. 2009, 58, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Thery, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, I.A.; Heuser, J.E.; Atkinson, J.P. GPI-anchored complement regulatory proteins in seminal plasma. An analysis of their physical conditions and the mechanisms of their binding to exogenous cells. J. Clin. Investig. 1996, 97, 1675–1686. [Google Scholar] [CrossRef] [Green Version]
- Kirchhoff, C.; Pera, I.; Derr, P.; Yeung, C.H.; Cooper, T. The molecular biology of the sperm surface. Post-testicular membrane remodelling. Adv. Exp. Med. Biol. 1997, 424, 221–232. [Google Scholar]
- Ekdahl, K.N.; Ronquist, G.; Nilsson, B.; Babiker, A.A. Possible immunoprotective and angiogenesis promoting roles for malignant cell-derived prostasomes: A new paradigm for prostatic cancer? Adv. Exp. Med. Biol. 2006, 586, 107–119. [Google Scholar]
- Babiker, A.A.; Ronquist, G.; Nilsson, U.R.; Nilsson, B. Transfer of prostasomal CD59 to CD59-deficient red blood cells results in protection against complement-mediated hemolysis. Am. J. Reprod. Immunol. 2002, 47, 183–192. [Google Scholar] [CrossRef]
- Babiker, A.A.; Nilsson, B.; Ronquist, G.; Carlsson, L.; Ekdahl, K.N. Transfer of functional prostasomal CD59 of metastatic prostatic cancer cell origin protects cells against complement attack. Prostate 2005, 62, 105–114. [Google Scholar] [CrossRef]
- Griffiths, G.S.; Galileo, D.S.; Reese, K.; Martin-Deleon, P.A. Investigating the role of murine epididymosomes and uterosomes in GPI-linked protein transfer to sperm using SPAM1 as a model. Mol. Reprod. Dev. 2008, 75, 1627–1636. [Google Scholar] [CrossRef]
- Griffiths, G.S.; Miller, K.A.; Galileo, D.S.; Martin-DeLeon, P.A. SPAM1 is secreted by the estrous murine uterus and oviduct in a form which can bind to sperm during capacitation: Acquisition enhances hyaluronic acid-binding ability and cumulus penetration efficiency. Reproduction 2008, 135, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Shen, J.; Wang, Y.; Pan, C.; Pang, W.; Diao, H.; Dong, W. Boar seminal plasma exosomes maintain sperm function by infiltrating into the sperm membrane. Oncotarget 2016, 7, 58832–58847. [Google Scholar] [CrossRef]
- Sullivan, R.; Saez, F.; Girouard, J.; Frenette, G. Role of exosomes in sperm maturation during the transit along the male reproductive tract. Blood Cells Mol. Dis. 2005, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rabesandrata, H.; Toutant, J.P.; Reggio, H.; Vidal, M. Decay-Accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood 1998, 91, 2573–2580. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Mainwaring, L.; Keyvanfar, K.; Chen, J.; Maciejewski, J.; Klein, H.G.; Young, N.S. Transfer of glycosylphosphatidylinositol-anchored proteins to deficient cells after erythrocyte transfusion in paroxysmal nocturnal hemoglobinuria. Blood 2004, 104, 3782–3788. [Google Scholar] [CrossRef]
- Anderson, S.M.; Yu, G.; Giattina, M.; Miller, J.L. Intercellular transfer of a glycosylphosphatidylinositol (GPI)-linked protein: Release and uptake of CD4-GPI from recombinant adeno-associated virus-transduced HeLa cells. Proc. Natl. Acad. Sci. USA 1996, 93, 5894–5898. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.L.; Giattina, M.; Mackie, E.J.; Dwyer, N.K. Variegated transfer of recombinant glycosylphosphatidylinositol-anchored CD4 among cultured cells: Correlation of flow cytometric and microscopic observations. J. Lab. Clin. Med. 1998, 131, 215–221. [Google Scholar] [CrossRef]
- Müller, G.; Schneider, M.; Gassenhuber, J.; Wied, S. Release of exosomes and microvesicles harbouring specific RNAs and glycosylphosphatidylinositol-anchored proteins from rat and human adipocytes is controlled by histone methylation. Am. J. Mol. Biol. 2012, 2, 187–209. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Jung, C.; Straub, J.; Wied, S.; Kramer, W. Induced release of membrane vesicles and exosomes from rat adipocytes containing lipid droplet, lipid raft and glycosylphosphatidylinositol-anchored proteins. Cell. Signal. 2009, 21, 324–338. [Google Scholar] [CrossRef]
- Müller, G.; Schneider, M.; Biemer-Daub, G.; Wied, S. Microvesicles released from rat adipocytes and harboring glycosylphosphatidylinositol-anchored proteins transfer RNA stimulating lipid synthesis. Cell. Signal. 2010, 23, 1207–1223. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Wied, S.; Biemer-Daub, G.; Frick, W. Transfer of the glycosylphosphatidylinositol-anchored 5′-nucleotidase CD73 from adiposomes into rat adipocytes stimulates lipid synthesis. Br. J. Pharmacol. 2010, 160, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Over, S.; Wied, S.; Frick, W. Association of cAMP-degrading glycosylphosphatidylinositol-anchored proteins with lipid droplets is induced by palmitate, H2O2 and the sulfonylurea drug, glimepiride, in rat adipocytes. Biochemistry 2008, 47, 12774–12787. [Google Scholar]
- Müller, G.; Wied, S.; Jung, C.; Over, S. Hydrogen peroxide-induced translocation of glycolipid-anchored cAMP-hydrolases to lipid droplets mediates inhibition of lipolysis in rat adipocytes. Br. J. Pharmacol. 2008, 154, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Wied, S.; Straub, J.; Jung, C. Coordinated regulation of esterification and lipolysis by palmitate, H2O2 and the anti-diabetic sulfonylurea drug, glimepiride, in rat adipocytes. Eur. J. Pharmacol. 2008, 597, 6–18. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Wied, S.; Biemer-Daub, G. Induced translocation of glycosylphosphatidylinositol-anchored proteins from lipid droplets to adiposomes in rat adipocytes. Br. J. Pharmacol. 2009, 158, 749–770. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Wied, S.; Walz, N.; Jung, C. Translocation of glycosylphosphatidylinositol-anchored proteins from plasma membrane microdomains to lipid droplets in rat adipocytes is induced by palmitate, H2O2 and the sulfonylurea drug, glimepiride. Mol. Pharmacol. 2008, 73, 1513–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Bosch, M.; Pol, A. Eukaryotic lipid droplets: Metabolic hubs, and immune first responders. Trends Endocrinol. Metab. 2022, 33, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Parton, R.G.; Pol, A. Lipid droplets, bioenergetic fluxes and metabolic flexibility. Semin. Cell Dev. Biol. 2020, 108, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Dejgaard, S.Y.; Presley, J.F. Interactions of lipid droplets with the intracellular transport machinery. Int. J. Mol. Sci. 2021, 22, 2776. [Google Scholar] [CrossRef]
- Jackson, C.L. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2019, 59, 88–96. [Google Scholar] [CrossRef]
- Müller, G.; Schneider, M.; Biemer-Daub, G.; Wied, S. Upregulation of lipid synthesis in small rat adipocytes by microvesicle-associated CD73 from large adipocytes. Obesity 2011, 19, 1531–1544. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Dearey, E.A.; Biemer-Daub, G. Glycosylphosphatidylinositol-anchored proteins coordinate lipolysis inhibition between large and small adipocytes. Metabolism 2011, 60, 1021–1037. [Google Scholar] [CrossRef] [PubMed]
- Müller, G. Let‘s shift lipid burden—From large to small adipocytes. Eur. J. Pharmacol. 2010, 656, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Rondinone, C.M. Adipocyte-derived hormones, cytokines, and mediators. Endocrine 2006, 29, 81–90. [Google Scholar] [CrossRef]
- Maslowska, M.; Wang, H.W.; Cianflone, K. Novel roles for acylation stimulating protein/C3ades/Arg: A review of recent in vitro and in vivo evidence. Vitam. Horm. 2005, 70, 309–332. [Google Scholar]
- Chou, W.L.; Chuang, L.M.; Chou, C.C.; Wang, A.H.; Lawson, J.A.; FitzGerald, G.A.; Chang, Z.F. Identification of a novel prostaglandin reductase reveals the involvement of E2 catabolism in regulation of peroxisome proliferator-activator receptor gamma activation. J. Biol. Chem. 2007, 282, 18162–18172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.H.; Chang, Y.C.; Su, T.H.; Nong, J.Y.; Li, C.C.; Chuang, L.M. Prostaglandin reductase-3 negatively modulates adipogenesis through regulation of PPARγ activity. J. Lipid Res. 2013, 54, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Basit, A.; Riaz, M.; Fawwad, A. Glimepiride: Evidence-based facts, trends, and observations. Vasc. Health Risk Manag. 2012, 8, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Tripathi, A.; Fanning, S.; Dettmer, U. Lipotoxicity downstream of a α-synuclein imbalance: A relevant pathomechanism in synucleinopathies. Biomolecules 2021, 12, 40. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and ß-cell failure in type 2 diabetes: Oxidative stress linked to NADPH oxidase and ER stress. Cells 2021, 10, 3328. [Google Scholar] [CrossRef]
- Benito-Vicente, A.; Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Uribe, K.B.; Martin, C. Molecular mechanisms of lipotoxicity-induced pancreatic ß-cell dysfunction. Int. Rev. Cell. Mol. Biol. 2021, 359, 357–402. [Google Scholar] [PubMed]
- Mastrototaro, L.; Roden, M. Insulin resistance and insulin sensitizing agents. Metabolism 2021, 125, 154892. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Salomon, D.S. Intercellular transfer regulation of the paracrine activity of GPI-anchored Cripto-1 as a Nodal co-receptor. Biochem. Biophys. Res. Commun. 2010, 403, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Hamada, S.; Bianco, C.; Mancino, M.; Nagaoka, T.; Gonzales, M.; Bailly, V.; Strizzi, L.; Salomon, D.S. Requirement of glycosylphosphatidylinositol anchor of Cripto-1 for trans activity as a Nodal co-receptor. J. Biol. Chem. 2007, 282, 35772–35786. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Nakayama, M.; Kawano, M.; Amagai, R.; Ishii, T.; Harigae, H.; Ogasawara, K. Fratricide of natural killer cells dressed with tumor-derived NKG2D ligand. Proc. Natl. Acad. Sci. USA 2013, 110, 9421–9426. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Cobo, S.; Romero-Cardenas, G.; Garcia-Cuesta, E.M.; Reyburn, H.T. Transfer of the human NKG2D ligands UL16 binding proteins (ULBP) 1-3 is related to lytic granule release and leads to ligand retransfer and killing of ULBP-recipient natural killer cells. Immunology 2015, 146, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.A.J.; Haldar, K.; Cross, G.A.M. Trypanosoma brucei variant surface glycoprotein has a sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. J. Biol. Chem. 1985, 260, 4963–4968. [Google Scholar] [CrossRef]
- Haldar, K.; Ferguson, M.A.J.; Cross, G.A.M. Acylation of a Plasmodium falciparum merozoite surface antigen via sn-1,2-diacyl glycerol. J. Biol. Chem. 1985, 260, 4969–4974. [Google Scholar] [CrossRef]
- Bouma, S.R.; Drislane, F.W.; Huestis, W.H. Selective extraction of membrane-bound proteins by phospholipid vesicles. J. Biol. Chem. 1977, 25, 6759–6763. [Google Scholar] [CrossRef]
- Newton, A.C.; Huestis, W.H. Vesicle-to-cell protein transfer: Insertion of band 3, the erythrocyte anion transporter, into lymphoid cells. Biochemistry 1988, 27, 4655–4659. [Google Scholar] [CrossRef]
- Huestis, W.H.; Newton, A.C. Intermembrane protein transfer: Band 3, the erythrocyte anion transporter, transfers in native orientation from human red blood cells into the bilayer of phospholipid vesicles. J. Biol. Chem. 1986, 261, 16274–16278. [Google Scholar] [CrossRef]
- Medof, M.E.; Kinoshita, T.; Nussenzweig, V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J. Exp. Med. 1984, 160, 1558–1578. [Google Scholar] [CrossRef]
- Medof, M.E.; Kinoshita, T.; Silber, R.; Nussenzweig, V. Amelioration of lytic abnormalities of paroxysmal nocturnal hemoglobinuria with decay-accelerating factor. Proc. Natl. Acad. Sci. USA 1985, 82, 2980–2984. [Google Scholar] [CrossRef] [Green Version]
- Low, M.G. Rapid intercellular transfer of GPI-anchored CD4. J. Lab. Clin. Med. 1998, 131, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Kooyman, D.L.; Byrne, G.W.; McClellan, S.; Nielsen, D.; Tone, M.; Waldmann, H.; Coffman, T.M.; McCurry, K.R.; Platt, J.L.; Logan, J.S. In Vivo Transfer of GPI-Linked Complement Restriction Factors from Erythrocytes to the Endothelium. Science 1995, 269, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Kooyman, D.L.; Byrne, G.W.; Logan, J.S. Glycosyl phosphatidylinositol anchor. Exp. Nephrol. 1998, 6, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Ilangumaran, S.; Robinson, P.J.; Hoessli, D.C. Transfer of exogenous glycosylphosphatidylinositol (GPI)-linked molecules to plasma membranes. Trends Cell Biol. 1996, 6, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.D.; Fukuoka, Y.; Sevlever, D.; Brunschwig, E.; Rosenberry, T.L.; Tykocinski, M.L.; Medof, M.E. Properties of exogenously added GPI-anchored proteins following their incorporation into cells. J. Cell. Biochem. 2001, 82, 234–245. [Google Scholar] [CrossRef]
- Kouzayha, A.; Besson, F. GPI-alkaline phosphatase insertion into phosphatidylcholine monolayers: Phase behavior and morphology changes. Biochem. Biophys. Res. Commun. 2005, 333, 1315–1321. [Google Scholar] [CrossRef]
- Suzuki, K.; Okumura, Y.; Sato, T. Membrane protein transfer from human erythrocyte ghosts to liposomes containing an artificial boundary lipid. Proc. Jpn. Acad. 1995, 71B, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Okumura, Y.; Sunamoto, J. Induction of acetylcholinesterase release from erythrocytes in the presence of liposomes. J. Biochem. 1999, 125, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Okumura, Y. GPI-linked proteins do not transfer spontaneously from erythocytes to liposomes. New aspects of reorganization of the cell membrane. Biochemistry 2000, 39, 9477–9485. [Google Scholar] [CrossRef]
- Suzuki, K.; Okumura, Y. Mechanism of selective release of membrane proteins from human erythrocytes in the presence of liposomes. Arch. Biochem. Biophys. 2000, 379, 344–352. [Google Scholar] [CrossRef]
- Medof, M.E.; Nagarajan, S.; Tykocinski, M.L. Cell-surface engineering with GPI-anchored proteins. FASEB J. 1996, 10, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Tykocinski, M.L.; Kaplan, D.R.; Medof, M.E. Antigen-presenting cell engineering. The molecular toolbox. Amer. J. Pathol. 1996, 148, 1–16. [Google Scholar]
- Rosse, W.F. New insights into paroxysmal nocturnal hemoglobinuria. Curr. Opin. Hematol. 2001, 8, 61–67. [Google Scholar] [CrossRef]
- Bessler, M.; Schaefer, A.; Keller, P. Paroxysmal nocturnal hemoglobinuria: Insights from recent advances in molecular biology. Transfus. Med. Rev. 2001, 15, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Medof, M.E.; Walter, E.I.; Roberts, W.L.; Haas, R.; Rosenberry, T.L. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry 1986, 25, 6740–6747. [Google Scholar] [CrossRef]
- Wilcox, L.A.; Ezzel, J.L.; Bernshaw, N.J.; Parker, C.J. Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood 1991, 78, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalman, L.S.; Wood, L.M.; Frank, M.M.; Müller-Eberhard, H.J. Deficiency of the homologous restriction factor in paroxysmal nocturnal hemoglobinuria. J. Exp. Med. 1987, 165, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, P.J. Phosphatidylinositol membrane anchors and T-cell activation. Immunol. Today 1991, 12, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J. Signal transduction via GPI-anchored membrane proteins. Adv. Exp. Med. Biol. 1997, 419, 365–370. [Google Scholar]
- Zhang, F.; Crise, B.; Su, B.; Hou, Y.; Rose, J.K.; Bothwell, A.; Jacobson, K. Lateral diffusion of membrane-spanning and glycosylphosphatidylinositol-linked proteins: Toward establishing rules governing the lateral mobility of membrane proteins. J. Cell Biol. 1991, 115, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Morgan, B.P.; van den Berg, C.W.; Davies, E.V.; Hallett, M.B.; Horejsi, V. Cross-linking of CD59 and of other glycosyl phosphatidylinositol-anchored molecules on neutrophils triggers cell activation via tyrosine kinase. Eur. J. Immunol. 1993, 23, 2841–2850. [Google Scholar] [CrossRef]
- Van den Berg, C.W.; Cinek, T.; Hallett, M.B.; Horejsi, V.; Morgan, B.P. Exogenous glycosyl phosphatidylinositol-anchored CD59 associates with kinases in membrane clusters on U937 cells and becomes Ca(2+)-signaling competent. J. Cell Biol. 1995, 131, 669–677. [Google Scholar] [CrossRef]
- Walter, E.I.; Ratnoff, W.D.; Long, K.E.; Kazura, J.W.; Medof, M.E. Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J. Biol. Chem. 1992, 267, 1245–1252. [Google Scholar] [CrossRef]
- Rifkin, M.R.; Landsberger, F.R. Trypanosome variant surface glycoprotein transfer to target membranes: A model for the pathogenesis of trypanosomiasis. Proc. Natl. Acad. Sci. USA 1990, 87, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Ilangumaran, S.; Arni, S.; Poincelet, M.; Theler, J.M.; Brennan, P.J.; Nasir-ud-Din; Hoessli, D.C. Integration of mycobacterial lipoarabinomannans into glycosylphosphatidylinositol-rich domains of lymphomonocytic cell plasma membranes. J. Immunol. 1995, 155, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Ilangumaran, S.; He, H.T.; Hoessli, D.C. Microdomains in lymphocyte signalling: Beyond GPI-anchored proteins. Immunol. Today 2000, 21, 2–7. [Google Scholar] [CrossRef]
- Nagarajan, S.; Anderson, M.; Ahmed, S.N.; Sell, K.W.; Selvaraj, P. Purification and optimization of functional reconstitution on the surface of leukemic cell lines of GPI-anchored Fc gamma receptor III. J. Immunol. Methods 1995, 184, 241–251. [Google Scholar] [CrossRef]
- Pearce, E.J.; Hall, B.F.; Sher, A. Host-spepcific evasion of the alternative complement pathway by schistosomes correlates with the presence of a phospholipase C-sensitive surface molecule resembling human decay accelerating factor. J. Immunol. 1990, 144, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- McCurry, K.R.; Kooyman, D.L.; Alvarado, C.G.; Cotterell, A.H.; Martin, M.J.; Logan, J.S.; Platt, J.L. Human complement regulatory proteins protect swine-to-primate cardiac xenografts from humoral injury. Nat. Med. 1995, 1, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Väkevä, A.; Lehto, T.; Takala, A.; Meri, S. Detection of a soluble form of the complement membrane attack complex inhibitor CD59 in plasma after acute myocardial infarction. Scand. J. Immunol. 2000, 52, 411–414. [Google Scholar] [CrossRef]
- Väkevä, A.; Jauhiainen, M.; Ehnholm, C.; Lehto, T.; Meri, S. High-density lipoproteins can act as carriers of glycophosphoinositol lipid-anchored CD59 in human plasma. Immunology 1994, 82, 28–33. [Google Scholar]
- Bütikofer, P.; Kuypers, F.A.; Xu, C.M.; Chiu, D.T.; Lubin, B. Enrichment of two glycosyl-phosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human red blood cells. Blood 1989, 74, 1481–1485. [Google Scholar] [CrossRef]
- Rooney, I.A.; Davies, A.; Morgan, B.P. Membrane attack complex (MAC)-mediated damage to spermatozoa: Protection of the cells by the presence on their membranes of MAC inhibitory proteins. Immunology 1992, 75, 499–506. [Google Scholar]
- Rooney, I.A.; Atkinson, J.P.; Krul, E.S.; Schonfeld, G.; Polakoski, K.; Saffitz, J.E.; Morgan, B.P. Physiologic relevance of the membrane attack complex inhibitory protein CD59 in human seminal plasma: CD59 is present on extracellular organelles (prostasomes), binds cell membranes, and inhibits complement-mediated lysis. J. Exp. Med. 1993, 177, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Vickram, A.S.; Samad, H.A.; Latheef, S.K.; Chakraborty, S.; Dhama, K.; Sridharan, T.B.; Sundaram, T.; Gulothungan, G. Human prostasomes as extracellular vesicles—Biomarkers for male infertility and prostate cancer: The journey from identification to current knowledge. Int. J. Biol. Macromol. 2020, 146, 946–958. [Google Scholar] [CrossRef]
- Sprong, H.; Suchanek, M.; van Dijk, S.M.; van Remoortere, A.; Klumperman, J.; Avram, D.; van der Linden, J.; Leusen, J.H.W.; van Hellemond, J.J.; Thiele, C. Abberrant receptor-mediated endocytosis of Schistosoma mansoni glycoproteins on host lipoproteins. PLoS Med. 2006, 3, e253. [Google Scholar] [CrossRef] [Green Version]
- Neumann, S.; Harterink, M.; Sprong, H. Hitch-hiking between cells on lipoprotein particles. Traffic 2007, 8, 331–338. [Google Scholar] [CrossRef]
- Niermann, T.; Kern, F.; Erne, P.; Resink, T. The glycosyl phosphatidylinositol anchor of human T-cadherin binds lipoproteins. Biochem. Biophys. Res. Commun. 2000, 276, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Panakova, D.; Sprong, H.; Marois, E.; Thiele, C.; Eaton, S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005, 435, 58–65. [Google Scholar] [CrossRef]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.S.; Galileo, D.S.; Aravindan, R.G.; Martin-DeLeon, P.A. Clusterin facilitates exchange of glycosyl phosphatidylinositol-linked SPAM1 between reproductive luminal fluids and mouse and human sperm membranes. Biol. Reprod. 2009, 81, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Martin-DeLeon, P.A. Germ-cell hyaluronidases: Their roles in sperm function. Int. J. Androl. 2011, 34, e306-18. [Google Scholar] [CrossRef]
- Martin-DeLeon, P.A. Epididymosomes: Transfer of fertility-modulating proteins to the sperm surface. Asian J. Androl. 2015, 17, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, R.; Pan, T.; Liu, D.; Petersen, R.B.; Wong, B.-S.; Gambetti, P.; Sy, M.S. Intercellular transfer of the cellular prion protein. J. Biol. Chem. 2002, 277, 47671–47678. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.A.; Ussar, S.; Tschöp, M.H.; Müller, T.D. Age-dependent membrane release and degradation of full-length glycosylphosphatidylinositol-anchored proteins in rats. Mech. Ageing Dev. 2020, 190, 111307. [Google Scholar] [CrossRef]
- Müller, G.A.; Lechner, A.; Tschöp, M.H.; Müller, T.D. Interaction of Full-Length Glycosylphosphatidylinositol-Anchored Proteins with Serum Proteins and Their Translocation to Cells In Vitro Depend on the (Pre-)Diabetic State in Rats and Humans. Biomedicines 2021, 9, 277. [Google Scholar] [CrossRef]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Chip-based sensing of the intercellular transfer of cell surface proteins: Regulation by the metabolic state. Biomedicines 2021, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Müller, T.D. Biological role of the intercellular transfer of glycosylphosphatidylinositol-anchored proteins: Stimulation of lipid and glycogen synthesis. Int. J. Mol. Sci. 2022, 23, 7418. [Google Scholar] [CrossRef]
- Izgilov, R.; Naftaly, A.; Benayahu, D. Advanced glycation end products effects on adipocyte niche stiffness and cell signaling. Int. J. Mol. Sci. 2023, 24, 2261. [Google Scholar] [CrossRef] [PubMed]
- Naftaly, A.; Kislev, N.; Izgilov, R.; Adler, R.; Silber, M.; Shalgi, R.; Benayahu, D. Nutrition alters the stiffness of adipose tissue and cell signaling. Int. J. Mol. Sci. 2022, 23, 15237. [Google Scholar] [CrossRef]
- Ben-Or, F.M.; Shoham, N.; Benayahu, D.; Gefen, A. Effects of accumulation of lipid droplets on load transfer between and within adipocytes. Biomech. Model Mechanobiol. 2015, 14, 15–28. [Google Scholar] [CrossRef]
- Goelzer, M.; Dudakovic, A.; Olcum, M.; Sen, B.; Ozcivici, E.; Rubin, J.; van Wijnen, A.J.; Uzer, G. Lamin A/C is dispensable to mechanical repression of adipogenesis. Int. J. Mol. Sci. 2021, 22, 6580. [Google Scholar] [CrossRef]
- Müller, G.A.; Herling, A.W.; Stemmer, K.; Lechner, A.; Tschöp, M.H. Chip-based sensing for release of unprocessed cell surface proteins in vitro and in serum and its (patho)physiological relevance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E212–E233. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Upregulated phospholipase D activity toward glycosylphosphatidylinositol-anchored proteins in micelle-like serum complexes in metabolically deranged rats and humans. Am. J. Physiol. Endocrinol. Metabol. 2020, 318, E462–E479. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Müller, T.D. Transfer of proteins from cultured human adipose to blood cells and induction of anabolic phenotype are controlled by serum, insulin and sulfonylurea drugs. Int. J. Mol. Sci. 2023, 24, 4825. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S. The sulfonylurea drug, glimepiride, stimulates glucose transport, glucose transporter translocation, and dephosphorylation in insulin-resistant rat adipocytes in vitro. Diabetes 1993, 42, 1852–1867. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Satoh, Y.; Geisen, K. Extrapancreatic effects of sulfonylureas—A comparison between glimepiride and conventional sulfonylureas. Diabetes Res. Clin. Pract. 1995, 28, S115–S137. [Google Scholar] [CrossRef]
- Frick, W.; Bauer, A.; Bauer, J.; Wied, S.; Müller, G. Structure-activity relationship of synthetic phosphoinositolglycans mimicking metabolic insulin action. Biochemistry 1998, 37, 13421–13436. [Google Scholar] [CrossRef]
- Huang, K.-S.; Li, S.; Fung, W.-J.C.; Hulmes, J.D.; Reik, L.; Pan, Y.-C.E.; Low, M.G. Purification and characterization of glycosyl-phosphatidylinositol-specific phospholipase D. J. Biol. Chem. 1990, 265, 17738–17745. [Google Scholar] [CrossRef]
- Stieger, S.; Diem, S.; Jakob, A.; Brodbeck, U. Enzymatic properties of phosphatidylinositol-glycan-specific phospholipase C from rat liver and phosphatidylinositol-glycan-specific phospholipase D from rat serum. Eur. J. Biochem. 1991, 197, 67–73. [Google Scholar] [CrossRef]
- Li, J.Y.; Hollfelder, K.; Huang, K.-S.; Low, M.G. Structural features of GPI-specific phospholipase D revealed by proteolytic fragmentation and Ca2+ binding studies. J. Biol. Chem. 1994, 269, 28963–28971. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, N.S.; Bowen, R.F.; Deeg, M.A. Mutating His29, His125, His133 or His158 abolishes glycosylphosphatidylinositol-specific phospholipase D catalytic activity. Biochem. J. 2005, 391, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, V.M.; Nelson, K.L.; Buckley, J.T.; Stevens, V.L.; Tweten, R.K.; Elwood, P.C.; Leppla, S.H. Clostridium septicum alpha toxin uses glycosylphosphatidylinositol-anchored protein receptors. J. Biol. Chem. 1999, 274, 27274–27280. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, S.; Hester, S.; Kirby, P.S.; Nairn, A.; Pierce, M.; Abbott, K.L. Elevated levels of glycosylphosphatidylinositol (GPI) anchored proteins in plasma from human cancers detected by C. septicum alpha toxin. Cancer Biomark. 2014, 14, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Varon Silva, D.; Lipowsky, R.; Santer, M. The importance of side branches of glycosylphosphatidylinositol anchors: A molecular dynamics perspective. Glycobiology 2022, 32, 933–948. [Google Scholar] [CrossRef]
- Kundu, S.; Lin, C.; Jaiswal, M.; Babu Mullapudi, V.; Craig, K.C.; Chen, S.; Guo, Z. Profiling glycosylphosphatidylinositol (GPI)-interacting proteins in the cell membrane using a bifunctional GPI analogue as the probe. J. Proteome Res. 2023, 22, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Babu Mullapudi, V.; Craig, K.C.; Guo, Z. Synthesis of a bifunctionalized glycosylphosphatidylinositol (GPI) anchor useful for the study of GPI biology. Chemistry 2022, 29, e202203457. [Google Scholar] [CrossRef]
- Guo, Z. Synthetic studies of glycosylphosphatidylinositol (GPI) anchors and GPI-anchored peptides, glycopeptides, and proteins. Curr. Org. Synth. 2013, 10, 366–383. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 2020, 369, 167–173. [Google Scholar] [CrossRef]
- Wieser, S.N.; Schnittger, L.; Florin-Christensen, M.; Delbecq, S.; Schetters, T. Vaccination against babesiosis using recombinant GPI-anchored proteins. Int. J. Parasitol. 2019, 49, 175–181. [Google Scholar] [CrossRef]
- Patel, J.M.; Kim, M.-C.; Vartabedian, V.F.; Lee, Y.-N.; He, S.; Song, J.-M.; Choi, H.-J.; Yamanaka, S.; Amaram, N.; Lukacher, A.; et al. Protein transfer-mediated surface engineering to adjuvanatate virus-like nanoparticles for enhanced anti-viral immune responses. Nanomedicine 2015, 11, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bumgarner, G.W.; Shashidharamurthy, R.; Nagarajan, S.; D’Souza, M.J.; Selvaraj, P. Surface engineering of microparticles by novel protein transfer for targeted antigen/drug delivery. J. Control. Release 2009, 137, 90–97. [Google Scholar] [CrossRef]
- McHugh, R.S.; Ahmed, A.N.; Wang, Y.C.; Sell, K.W.; Selvaraj, P. Construction, purification and functional reconstitution on tumor cells of a glycolipid anchored human B7-1 (CD80). Proc. Natl. Acad. Sci. USA 1995, 92, 8059–8063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, R.S.; Nagarajan, S.; Wang, Y.C.; Sell, K.W.; Selvaraj, P. Protein transfer of glycosyl-phosphatidylinositol-B7-1 into tumor cell membranes: A novel approach to tumor immunotherapy. Cancer Res. 1999, 59, 2433–2437. [Google Scholar]
- Cimino, A.M.; Palaniswarni, P.; Kim, A.C.; Selvaraj, P. Cancer vaccine development: Protein transfer of membrane-anchored cytokines and immunostimulatory molecules. Immunol. Res. 2004, 29, 231–240. [Google Scholar] [CrossRef]
- Patel, J.M.; Vartabedian, V.F.; Kim, M.-C.; He, S.; Kang, S.-M.; Sevaraj, P. Influenza virus-like particles engineered by protein transfer with tumor-associated antigens induces protective antitumor immunity. Biotechnol. Bioeng. 2015, 112, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.; Selvaraj, P. Expression and characterization of glycolipid-anchored B7-1 (CD80) from baculovirus-infected insect cells: Protein transfer onto tumor cells. Protein Expr. Purif. 1999, 17, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bommireddy, R.; Munoz, L.E.; Kuman, A.; Huang, L.; Fan, Y.; Monterroza, L.; Pack, C.D.; Ramachandiran, S.; Reddy, S.J.C.; Kim, J.; et al. Tumor membrane vesicle vaccine augments the efficacy of anti-PD1 antibody in immune checkpoint inhibitor-resistant squamous cell carcinoma models of head and neck cancer. Vaccines 2020, 8, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunschwig, E.B.; Fayen, J.D.; Medof, M.E.; Tykocinski, M.L. Protein transfer of glycosyl-phosphatidylinositol (GPI)-modified murine B7-1 and B7-2 costimulators. J. Immunother. 1999, 22, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.M.; Vartabedian, V.F.; Bozeman, E.N.; Caoyonan, B.E.; Srivatsan, S.; Pack, C.D.; Dey, P.; D’Souza, M.J.; Yang, L.; Selvaraj, P. Plasma membrane vesicles decorated with glycolipid-anchored antigens and adjuvants via protein transfer as an antigen delivery platform for inhibition of tumor growth. Biomaterials 2016, 74, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, S.; Selvaraj, P. Human tumor membrane vesicles modified to express glycolipid-anchored IL-12 by protein transfer induce T cell proliferation in vitro: A potential approach for local delivery of cytokines during vaccination. Vaccine 2006, 24, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Getty, R.R.; Chisari, F.V.; Fowler, P.; Greenspan, N.S.; Tykocinski, M.L. Protein transfer of preformed MHC-peptide complexes sensitizes target cells to T cell cytolysis. Immunity 1994, 1, 607–613. [Google Scholar] [CrossRef]
- Zhao, X.; Li, H.; Lee, R.J. Targeted drug delivery via folate receptors. Expert Opin. Drug Deliv. 2008, 5, 309–319. [Google Scholar] [CrossRef]
- Stachel, G.; Trenkwalder, T.; Götz, F.; Aouni, C.E.; Muenchmeier, N.; Pfosser, A.; Nussbaum, C.; Sperandio, M.; Hatzopoulos, A.K.; Hinkel, R.; et al. SDF-1 fused to a fractalkine stalk and a GPI anchor enables functional neovascularization. Stem Cells 2013, 31, 1795–1805. [Google Scholar] [CrossRef]
- Dunn, D.E.; Yu, J.; Nagarajan, S.; Devetten, M.; Weichold, F.F.; Medof, M.E.; Young, N.S.; Liu, J.M. A knock-out model of paroxysmal nocturnal hemoglobinuria: Pig-a(-) hematopoiesis is reconstituted following intercellular transfer of GPI-anchored proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 7938–7943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloand, E.M.; Maciejewski, J.P.; Dunn, D.; Moss, J.; Brewer, B.; Kirby, M.; Young, N.S. Correction of the PNH defect by GPI-anchored protein transfer. Blood 1998, 92, 4439–4445. [Google Scholar] [CrossRef]
- Müller, G. Microvesicles/exosomes as potential novel biomarkers of metabolic diseases. Diabetes Metab. Syndr. Obes. 2012, 5, 247–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, L.; Vassali, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Salunkhe, S.; Dheeraj; Basak, M.; Chitkara, D.; Mittal, A. Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: Strategies and significance. J. Control. Release 2020, 326, 599–614. [Google Scholar] [PubMed]
- Tykocinski, M.L.; Chen, A.; Huang, J.-H.; Weber, M.C.; Zheng, G. New designs for cancer vaccine and artificial veto cells: An emerging palette of protein paints. Immunol. Res. 2003, 27, 565–574. [Google Scholar] [CrossRef]
- Nishimura, J.; Smith, C.A.; Phillips, K.L.; Ware, R.E.; Rosse, W.F. Paroxysmal nocturnal hemoglobinuria: Molecular pathogenesis and molecular therapeutic approaches. Hematopathol. Mol. Hematol. 1998, 11, 119–146. [Google Scholar] [PubMed]
- Brodsky, R.A.; Jane, S.M.; Vanin, E.F.; Mitsuya, H.; Peters, T.R.; Shimada, T.; Medof, M.E.; Nienhus, A.W. Purified GPI-anchored CD4DAF as a receptor for HIV-mediated gene transfer. Hum. Gene Ther. 1994, 5, 1231–1239. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Shai, N.; Schuldiner, M.; Bohnert, M. A tether is a tether is a tether: Tethering at membrane contact sites. Dev. Cell 2016, 39, 395–409. [Google Scholar] [CrossRef]
- Jain, A.; Holthuis, J.C.M. Membrane contact sites, ancient and central hubs of cellular lipid logistics. Biochim. Biophys. Acta 2017, 1864, 1450–1458. [Google Scholar] [CrossRef]
- Caro, L.H.; Tettelin, H.; Vossen, J.H.; Ram, A.F.; van den Ende, H.; Klis, F.M. In silico identification of glycosyl-phosphatidylinositol-anchored plasma-membrane and cell wall proteins of Saccharomyces cerevisiae. Yeast 1997, 13, 1477–1489. [Google Scholar] [CrossRef]
- Kapteyn, J.C.; Van Den Ende, H.; Klis, F.M. The contribution of cell wall proteins to the organisation of the yeast cell wall. Biochim. Biophys. Acta 1999, 1426, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Klis, F.M.; Boorsma, A.; De Groot, P.W. Cell wall construction in Saccharomyces cerevisiae. Yeast 2006, 23, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Shimoi, H.; Iimura, Y. Structure of the glucan-binding sugar chain of Tip1p, a cell wall protein of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1999, 1427, 133–144. [Google Scholar] [CrossRef]
- Popolo, L.; Vai, M. The Gas1 glycoprotein, a putative wall polymer cross-linker. Biochim. Biophys. Acta 1999, 1426, 385–400. [Google Scholar] [CrossRef]
- Müller, G.; Groß, E.; Wied, S.; Bandlow, W. Glucose-induced sequential processing of a glycosyl-phosphatidylinositol-anchored ectoprotein in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 442–456. [Google Scholar] [CrossRef] [Green Version]
- van der Vaart, J.M.; Caro, L.H.; Chapman, J.W.; Klis, F.M.; Verrips, C.T. Identification of three mannoproteins in the cell wall of Saccharomyces cerevisiae. J. Bacteriol. 1995, 177, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Vossen, J.H.; Muller, W.H.; Lipke, P.N.; Klis, F.M. Restrictive glycosylphosphatidylinositol anchor synthesis in cwh6/gpi3 yeast cells causes aberrant biogenesis of cell wall proteins. J. Bacteriol. 1997, 179, 2202–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, K.; Terashima, H.; Arisawa, M.; Kitada, K. Amino acid residues in the omega-minus region participate in cellular localization of yeast glycosylphosphatidylinositol-attached proteins. J. Bacteriol. 1999, 181, 3886–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoko-o, T.; Umemura, M.; Komatsuzaki, A.; Ikeda, K.; Ichikawa, D.; Takase, K.; Kanzawa, N.; Saito, K.; Kinoshita, T.; Taguchi, R.; et al. Lipid moiety of glycosylphosphatidylinositol-anchored proteins contributes to the determination of their final destination in yeast. Genes Cells 2018, 23, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Kitagaki, H.; Wu, H.; Shimoi, H.; Ito, K. Two homologous genes, DCW1 (YKL046c) and DFG5, are essential for cell growth and encode glycosylphosphatidylinositol (GPI)-anchored membrane proteins required for cell wall biogenesis in Saccharomyces cerevisiae. Mol. Microbiol. 2002, 46, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- de Sampaio, G.; Bourdineaud, J.P.; Lauquin, G.J. A constitutive role for GPI anchors in Saccharomyces cerevisiae: Cell wall targeting. Mol. Microbiol. 1999, 34, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Yoko-o, T.; Ichikawa, D.; Miyagishi, Y.; Kato, A.; Umemura, M.; Takase, K.; Ra, M.; Ikeda, K.; Taguchi, R.; Jigami, Y. Determination and physiological roles of the glycosylphosphatidylinositol lipid remodelling pathway in yeast. Mol. Microbiol. 2013, 88, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Seals, J.R.; Jarett, L. Activation of pyruvate dehydrogenase by direct addition of insulin to an isolated plasma membrane/mitochondria mixture: Evidence for generated of insulin’second messenger in a subcellular system. Proc. Natl. Acad. Sci. USA 1980, 77, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Wied, S.; Crecelius, A.; Kessler, A.; Eckel, J. Phosphoinositolglycan-peptides from yeast potently induce metabolic insulin actions in isolated rat adipocytes, cardiomyocytes, and diaphragms. Endocrinology 1997, 138, 3459–3475. [Google Scholar] [CrossRef]
- Gupta, A.; Jakubowicz, D.; Nestler, J.E. Pioglitazone therapy increases insulin-stimulated release of d-chiro-inositol-containing inositolphosphoglycan mediator in women with polycystic ovary syndrome. Metab. Syndr. Relat. Disord. 2016, 14, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Stralfors, P. Insulin second messengers. Bioessays 1997, 19, 327–335. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Cuatrecasas, P. In search of a second messenger for insulin. Am. J. Physiol. 1988, 255, C1–C11. [Google Scholar] [CrossRef]
- Varela-Nieto, I.; Leon, Y.; Caro, H.N. Cell signalling by inositol phosphoglycans from different species. Comp. Biochem. Physiol. Biochem. Mol. Biol. 1996, 115, 223–241. [Google Scholar] [CrossRef]
- Larner, J.; Huang, L.C.; Tang, G.; Suzuki, S.; Schwartz, C.F.; Romero, G.; Roulidis, Z.; Zeller, K.; Shen, T.Y.; Oswald, A.S. Insulin mediators: Structure and formation. Cold Spring Harb. Symp. Quant. Biol. 1988, 53, 965–971. [Google Scholar] [CrossRef]
- Hecht, M.-L.; Tsai, Y.-H.; Liu, X.; Wolfrum, C.; Seeberger, P.H. Synthetic inositol phosphoglycans related to GPI lack insulin-mimetic activity. ACS Chem. Biol. 2010, 5, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Luo, M.; To, K.K.W.; Zhang, J.; Su, C.; Zhang, H.; An, S.; Wang, F.; Chen, D.; Fu, L. Intercellular transfer of exosomal wild type EGFR triggers Osimertinib resistance in non-small cell lung cancer. Mol. Cancer 2021, 20, 17. [Google Scholar] [CrossRef]
- Huang, D.; Chen, J.; Hu, D.; Xie, F.; Yang, T.; Li, Z.; Wang, X.; Xiao, Y.; Zhong, J.; Jiang, Y.; et al. Advances in biological function and clinical application of small extracellular vesicle membrane proteins. Front. Oncol. 2021, 11, 675940. [Google Scholar] [CrossRef]
- Hargett, L.A.; Bauer, N.N. On the origin of microparticles: From “platelet dust” to mediators of intercellular communication”. Pulm. Circ. 2013, 3, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, Y.; Buzas, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Thery, C.; et al. A brief history of nearly EV-erything—The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Zhang, N.; Hu, X.; Wang, H. Tumor-associated exosomes promote lung cancer metastasis through multiple mechanisms. Mol. Cancer 2021, 20, 117. [Google Scholar] [CrossRef]
- Liu, Q.; Piao, H.; Wang, Y.; Zheng, D.; Wang, W. Circulating exosomes in cardiovascular disease: Novel carriers of biological information. Biomed. Pharmacother. 2021, 135, 111148. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. Information transfer by exosomes: A new frontier in hematologic malignancies. Blood Rev. 2015, 29, 281–290. [Google Scholar] [CrossRef]
- You, J.X.; Qi, S.N.; Fu, J.L.; Wang, C.G.; Su, G.F. Circulating exosomes in ophthalmic disease: Novel carriers of biological information. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2172–2181. [Google Scholar]
- Sasaki, R.; Kanda, T.; Yokosuka, O.; Kato, N.; Matsuoka, S.; Moriyama, M. Exosomes and hepatocellular carcinoma: From bench to bedside. Int. J. Mol. Sci. 2019, 20, 1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.; Garikipati, V.N.S.; Gumpert, A. Tiny shuttles for information transfer: Exosomes in cardiac health and disease. J. Cardiovasc. Transl. Res. 2016, 9, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Science 2000, 113, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C.; Cook, S.L.; Huestis, W.H. Transfer of band 3, the erythrocyte anion transporter, between phospholipid vesicles and cells. Biochemistry 1983, 22, 6110–6117. [Google Scholar] [CrossRef]
- Hertz, L.; Flormann, D.; Birnbaumer, L.; Wagner, C.; Laschke, M.W.; Kaestner, L. Evidence of in vivo exogen protein uptake by red blood cells: A putative therapeutic concept. Blood Advanc. 2023, 7, 1033–1039. [Google Scholar] [CrossRef]
- Dance, A. Core concept: Cells nibble one another via the under-appreciated process of trogocytosis. Proc. Natl. Acad. Sci. USA 2019, 116, 17608–17610. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Karasuyama, H. The role of trogocytosis in the modulation of immune cell functions. Cells 2021, 10, 1255. [Google Scholar] [CrossRef]
- Reed, J.; Reichelt, M.; Wetzel, S.A. Lymphocytes and trogocytosis-mediated signaling. Cells 2021, 10, 1478. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Commun. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopedia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. 2019, 27, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.K.; Ruthazer, E.S. Microglial trogocytosis and the complement system regulate axonal pruning in vivo. eLife 2021, 10, e62167. [Google Scholar] [CrossRef]
- Vanherberghen, B.; Andersson, K.; Carlin, L.M.; Nolte-t Hoen, E.N.; Williams, G.S.; Hoglund, P.; Davis, D.M. Human and murine inhibitory natural killer cell receptors transfer from natural killer cells to target cells. Proc. Natl. Acad. Sci USA 2004, 101, 16873–16878. [Google Scholar] [CrossRef] [Green Version]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houndt, M.; Treffers, L.W.; van Rees, D.S.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils kill antibody-opsonized cancer cells by trogocytosis. Cell Rep. 2018, 23, 3946–3959. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Balatskaya, M.N.; Tkachuk, V.A. Glycosylphosphatidylinositol-anchored proteins as regulators of cortical cytoskeleton. Biochemistry 2016, 81, 636–650. [Google Scholar] [CrossRef]
- Kalappurakkal, J.M.; Sil, P.; Mayor, S. Toward a new picture of the living plasma membrane. Protein Sci. 2020, 29, 1355–1365. [Google Scholar] [CrossRef]
- Jacobson, K.; Liu, P.; Lagerholm, B.C. The lateral organization and mobility of plasma membrane components. Cell 2019, 177, 806–819. [Google Scholar] [CrossRef]
- Saltukoglu, D.; Özdemir, B.; Holtmannspötter, M.; Reski, R.; Piehler, J.; Kurre, R.; Reth, M. Plasma membrane topography governs the 3D dynamic localization of IgM B cell antigen receptor clusters. EMBO J. 2023, 42, e112030. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R. The Doctrine of DNA: Biology as Ideology; Penguin: London, UK, 1993. [Google Scholar]
- Lewontin, R. The Triple Helix: Gene, Organism and Environment; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Griffith, F. The significance of pneumococcal types. J. Hyg. 1928, 27, 113–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, F.H.C. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Kay, L.E. Who Wrote the Book of Life?: A History of the Genetic Code; Stanford University Press: Stanford, CA, USA, 2000. [Google Scholar]
- Barad, K. Agential realism: How material-discursive practices matter. Signs 2003, 28, 803–831. [Google Scholar]
- Kerr, A. Genetics and Society: A Sociology of Disease; Routledge: London, UK, 2004. [Google Scholar]
- Rheinberger, H.-J. Iterationen; Merve Press: Berlin, Germany, 2005. [Google Scholar]
- Fairbanks, D.J. Mendel and Darwin: Untangling a persistent enigma. Heredity 2020, 124, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, P.J.; Noel Ellis, T.H. Mendel’s reaction of Darwin’s provisional hypothesis of pangenesis and the experiment that could not wait. Heredity 2022, 129, 12–16. [Google Scholar] [CrossRef]
- Barad, K. Meeting the Universe Halfway; Duke University Press: Durham, NC, USA; London, UK, 2007. [Google Scholar]
- Berman, J.D.; Young, M. Rapid and complete purification of acetylcholinesterases of electric eel and erythrocyte by affinity chromatography. Proc. Natl. Acad. Sci. USA 1971, 68, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Toutant, J.P.; Roberts, W.L.; Murray, N.R.; Rosenberry, T.L. Conversion of human erythrocyte acetylcholinesterase from an amphiphilic to a hydrophilic form by phosphatidylinositol-specific phospholipase C and serum phospholipase D. Eur. J. Biochem. 1989, 180, 503–508. [Google Scholar] [CrossRef]
- Müller, G.; Jordan, H.; Petry, S.; Wetekam, E.-M.; Schindler, P. Analysis of lipid metabolism in adipocytes using fluorescent fatty acids. I. Insulin stimulation of lipogenesis. Biochim. Biophys. Acta 1997, 1347, 23–39. [Google Scholar] [CrossRef]
- Pinto Correia, C. The Ovary of Eve: Egg and Sperm and Preformation; University of Chicago Press: Chicago, IL, USA, 2007. [Google Scholar]
- Benson, K. Epigenesis. In The Oxford Encyclopedia of Evolution; Pagel, M., Ed.; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- De Lamarck, J.B. Philosophie Zoologique; Musée d‘Histoire Naturelle: Paris, France, 1809. [Google Scholar]
- Weismann, A. Essays upon Heredity and Kindred Biological Problems; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Weismann, A. Das Keimplasma: Eine Theorie der Vererbung; Gustav Fischer: Stuttgart, Germany, 1892. [Google Scholar]
- Collins, H.M.; Yearly, S. Epistemological Chicken. In Science as Practice and Culture; Pickering, A., Ed.; University of Chicago Press: Chicago, IL, USA; London, UK, 1992; pp. 301–326. [Google Scholar]
- Hacking, I. Representing and Intervening; Cambridge University Press: Cambridge, MA, USA, 1983. [Google Scholar]
- Pickering, A. The Mangle of Practice: Time, Agency, and Science; University of Chicago Press: Chicago, IL, USA, 1995. [Google Scholar]
- Rouse, J. Engaging Science: How to Understand Its Practices Philosophically; Cornell University Press: Itacha, NY, USA, 1996. [Google Scholar]
- Callon, M.; Latour, B. Don’t Throw the Baby out with the Bath School! A Reply to Collins and Yearly. In Science as Practice and Culture; Pickering, A., Ed.; University of Chicago Press: Chicago, IL, USA; London, UK, 1992; pp. 343–368. [Google Scholar]
- Latour, B. We Have Never Been Modern; Harvard University Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Bohr, N. Atomic Physics and Human Knowledge; John Wiley and Sons Inc.: New York, NY, USA, 1958. [Google Scholar]
- Barad, K. Getting real: Technoscientific practices and the materialization of reality. Differ. J. Fem. Cult. Stud. 1998, 10, 87–126. [Google Scholar] [CrossRef]
- Barad, K. Posthumanist performativity: How matter comes to matter. Signs J. Women Cult. Sci. 2003, 28, 801–831. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.A.; Müller, T.D. (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters. Biomolecules 2023, 13, 994. https://doi.org/10.3390/biom13060994
Müller GA, Müller TD. (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters. Biomolecules. 2023; 13(6):994. https://doi.org/10.3390/biom13060994
Chicago/Turabian StyleMüller, Günter A., and Timo D. Müller. 2023. "(Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters" Biomolecules 13, no. 6: 994. https://doi.org/10.3390/biom13060994
APA StyleMüller, G. A., & Müller, T. D. (2023). (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters. Biomolecules, 13(6), 994. https://doi.org/10.3390/biom13060994