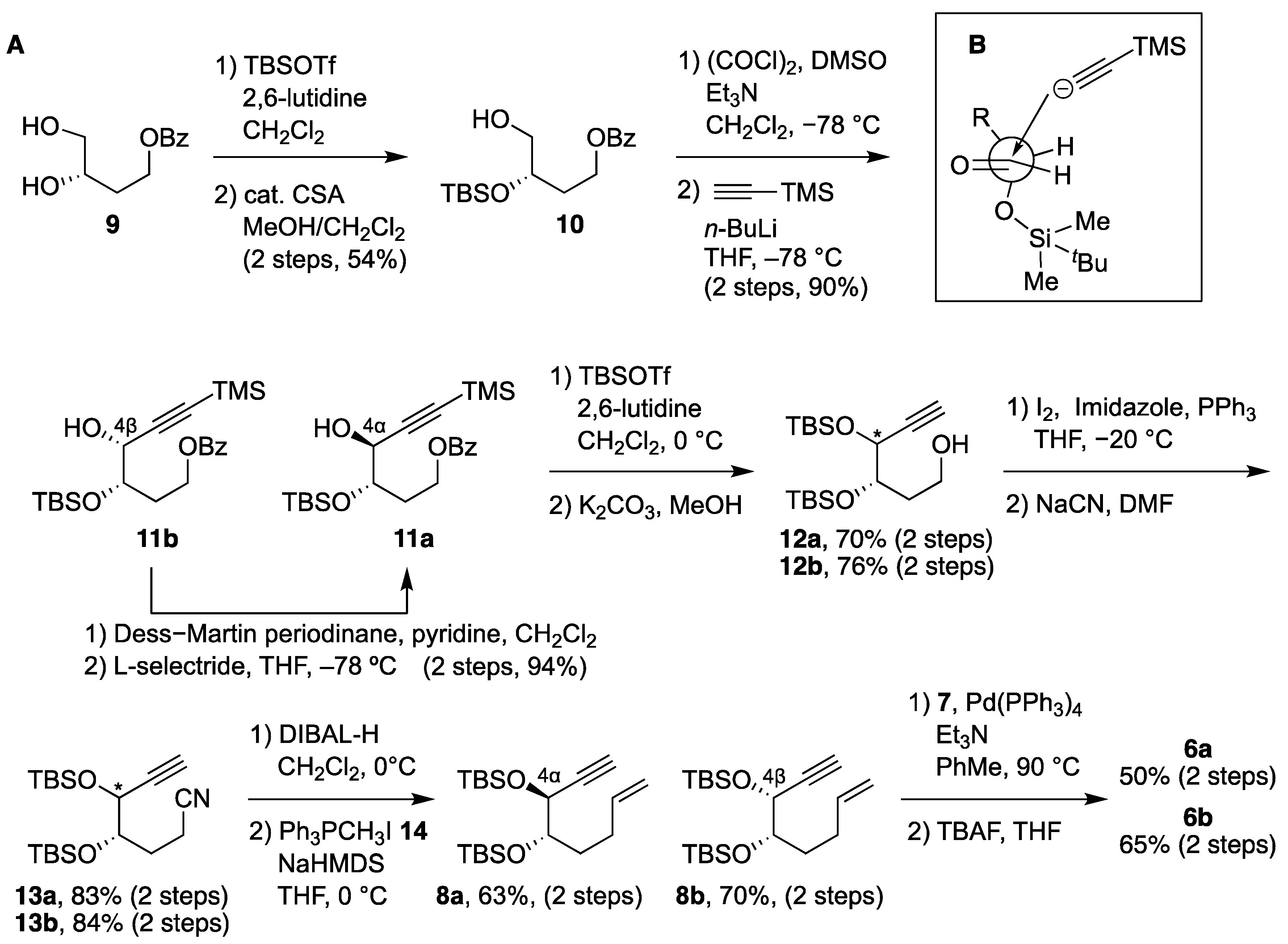
2.1. Synthesis of 6a and 6b
All reagents were supplied by commercial sources without further purification. All reactions involving air- or moisture-sensitive reagents were carried out in flame-dried glassware under argon atmosphere. Flash chromatography was performed using silica gel 60 (spherical, particle size 0.040−0.100 mm; Kanto Co., Inc., Tokyo, Japan), and preparative TLC (PLC) was performed using PLC silica gel 60 F254 (0.5 mm, Merck Ltd., Darmstadt, Germany). Optical rotations were measured on a JASCO P-2200 polarimeter (JASCO Co., Inc., Tokyo, Japan). 1H and 13C NMR spectra were recorded on JNM-AL300 (300 MHz, JEOL Ltd., Tokyo, Japan), JNM-ECX 400 (400 MHz, JEOL Ltd., Tokyo, Japan), and JNM-ECA 500 (500 MHz, JEOL Ltd., Tokyo, Japan) spectrometers. Chemical shift in CDCl3 was reported in the scale relative to CDCl3 (7.26 ppm) for 1H NMR. For 13C NMR, the chemical shift was reported in the scale relative to CDCl3 (77.0 ppm) and CD3OD (49.0 ppm) as an internal reference. HRMS (ESI) measurements were performed on a JMS-T100LC spectrometer (JEOL Ltd., Tokyo, Japan).
(
S)-3,4-bis((
tert-butyldimethylsilyl)oxy)butyl benzoate (
S1): To a solution of diol
9 [
13] (300 mg, 1.4 mmol) in CH
2Cl
2 (7 mL) was added 2,6-lutidine (0.44 mL, 3.7 mmol) and
tert-butyldimethylsilyl triflate (0.82 mL, 3.6 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H
2O, and the aqueous layer was extracted with CH
2Cl
2 three times. The combined organic layer was washed with brine, dried over MgSO
4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (
n-hexane/ethyl acetate = 20:1) to give the protected product
S1 (627 mg, 99%) as a colorless oil. [α]
= –10.6 (
c 0.87, CHCl
3);
1H NMR (300 MHz, CDCl
3) δ 8.05 (d,
J = 7.9 Hz, 2H), 7.55 (t,
J = 7.7 Hz, 1H), 7.44 (t,
J = 7.7 Hz, 2H), 4.34–4.51 (m, 2H), 3.86–3.94 (m, 1H), 3.62 (q,
J = 4.9 Hz, 1H), 3.48 (q,
J = 5.5 Hz, 1H), 2.05–2.15 (m, 1H), 1.81 (td,
J = 13.6, 5.6 Hz, 1H), 0.89 (s, 18H), 0.07 (d,
J = 4.8 Hz, 12H);
13C NMR (75 MHz, CDCl
3) δ 166.5, 132.8, 130.4, 129.5, 128.3, 70.1, 67.5, 61.8, 33.4, 31.1, 25.9, 25.8, 25.7, −3.0, −4.3, −5.0, −5.4; HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
23H
42O
4Si
2Na 461.2519, found 461.2493.
(S)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxybutyl benzoate (10): To a solution of diol S1 (627 mg, 1.4 mmol) in MeOH/THF = 1:1 (17 mL) was added (±)-CSA (40 mg, 0.17 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give 10 (77 mg, 54%) as a colorless oil. [α] = –9.8 (c 0.53, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 4.31–4.49 (m, 2H), 3.96–4.03 (m, 1H), 3.66 (q, J = 4.9 Hz, 1H), 3.55 (q, J = 5.3 Hz, 1H), 1.99 (q, J = 6.3 Hz, 2H), 0.91 (s, 9H), 0.10 (d, J = 2.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.9, 130.2, 129.5, 128.4, 69.7, 66.4, 61.6, 32.9, 25.8, 18.0, −4.6, −4.8; HRMS (ESI) m/z: [M + Na]+ calcd for C17H28O4SiNa 347.1655, found 347.1670.
(3S,4R)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (11b): To a solution of oxalic acid dichloride (0.48 mL, 5.6 mmol) in CH2Cl2 (7 mL) was added dimethyl sulfoxide (1.0 mL, 14.0 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 10 min, then a solution of 10 in CH2Cl2 (0.4 M, 7 mL, 2.78 mmol) and triethylamine (3.9 mL, 27.8 mmol) were added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 1:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of trimethylsilylacetylene (1.0 mL, 7.2 mmol) in THF (12 mL) was added n-butyllithium (2.6 M in hexane; 2.3 mL, 6.0 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.3 M, 9.3 mL, 2.78 mmol) was added dropwise at the same temperature. The resulting mixture was stirred at the same temperature for 15 min. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 11b (910 mg, 90%) as a colorless oil. [α] = 1.5 (c 5.36, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 6.9 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 4.48–4.56 (m, 1H), 4.30–4.42 (m, 2H), 3.99–4.04 (m, 1H), 2.16–2.27 (m, 1H), 1.97–2.08 (m, 1H), 0.91 (s, 9H), 0.16 (s, 9H), 0.10 (d, J = 5.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 166.4, 132.9, 130.2, 129.5, 128.3, 103.0, 91.7, 71.9, 66.5, 61.6, 31.1, 25.7, 18.0, −0.3, −4.5, −4.7; HRMS (ESI) m/z: [M + Na]+ calcd for C22H36O4Si2Na 443.2050, found 443.2017.
(S)-3-((tert-butyldimethylsilyl)oxy)-4-oxo-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S2): To a solution of 11b (100 mg, 0.24 mmol) and pyridine (0.38 mL) in CH2Cl2 (19 mL) was added Dess–Martin periodinane (404 mg, 0.95 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give S2 (94 mg, 94%) as a colorless oil. [α] = +0.7 (c 1.37, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 6.9 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.4 Hz, 2H), 4.46–4.56 (m, 1H), 4.32–4.43 (m, 2H), 2.18–2.26 (m, 2H), 0.93 (s, 9H), 0.05–0.22 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 189.5, 166.3, 132.9, 130.0, 129.6, 128.3, 102.4, 100.4, 75.5, 60.1, 33.7, 31.1, 25.7, 18.1, −1.0, −4.7, −5.4; HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O4Si2Na 441.1893, found 441.1877.
(3S,4S)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (11a): To a solution of S2 (94 mg, 0.22 mmol) in THF (11 mL) was added L-selectride (1.0 M in THF, 0.40 mL, 0.40 mmol) at –78 °C. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give 11a (94 mg, 99%) as a colorless oil. [α] = –9.1 (c 0.35, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 6.9 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 4.32–4.51 (m, 3H), 3.99 (q, J = 5.4 Hz, 1H), 1.99–2.19 (m, 2H), 0.92 (s, 9H), 0.12–0.16 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 166.4, 132.9, 130.1, 129.5, 128.3, 104.6, 90.6, 72.5, 65.8, 61.2, 32.7, 25.9, 18.1, −0.3, −4.3, −4.6; HRMS (ESI) m/z: [M + Na]+ calcd for C22H36O4Si2Na 443.2050, found 443.2054.
(3S,4S)-3,4-bis((tert-butyldimethylsilyl)oxy)-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S3a): To a solution of 11a (115 mg, 0.27 mmol) in CH2Cl2 (3 mL) was added 2,6-lutidine (98 μL, 0.82 mmol) and tert-butyldimethylsilyl triflate (94 μL, 0.41 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 40:1) to give S3a (143 mg, 98%) as a colorless oil. [α] = –5.6 (c 0.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.2 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 4.35–4.58 (m, 3H), 3.81–3.87 (m, 1H), 2.14–2.25 (m, 1H), 1.95–2.09 (m, 1H), 0.90 (d, J = 1.4 Hz, 18H), 0.05–0.16 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.8, 130.5, 129.5, 128.3, 104.8, 90.5, 71.5, 67.3, 61.8, 31.2, 25.8, 25.8, 18.2, 18.0, −0.3, −4.5, −4.6, −4.8, −4.9; HRMS (ESI) m/z: [M + Na]+ calcd for C28H50O4Si3Na 557.2915, found 557.2909.
(3S,4R)-3,4-bis((tert-butyldimethylsilyl)oxy)-6-(trimethylsilyl)hex-5-yn-1-yl benzoate (S3b): To a solution of 11b (264 mg, 0.63 mmol) in CH2Cl2 (6 mL) was added 2,6-lutidine (0.22 mL, 1.9 mmol) and tert-butyldimethylsilyl triflate (0.22 mL, 0.94 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 40:1) to give S3b (334 mg, 99%) as a colorless oil. [α] = –26.2 (c 1.38, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.4 Hz, 2H), 4.46–4.54 (m, 1H), 4.36–4.42 (m, 1H), 4.28 (d, J = 4.5 Hz, 1H), 3.89–3.94 (m, 1H), 2.08–2.19 (m, 1H), 1.92–2.05 (m, 1H), 0.91 (d, J = 1.4 Hz, 18H), 0.05–0.15 (m, 21H); 13C NMR (75 MHz, CDCl3) δ 166.5, 132.8, 130.5, 129.5, 128.3, 105.8, 90.3, 72.9, 67.8, 61.8, 32.0, 25.9, 25.8, 18.1, −0.3, −4.1, −4.4, −4.8, −5.0; HRMS (ESI) m/z: [M + Na]+ calcd for C28H50O4Si3Na 557.2915, found 557.2872.
(3S,4S)-3,4-bis((tert-butyldimethylsilyl)oxy)hex-5-yn-1-ol (12a): To a solution of S3a (143 mg, 0.27 mmol) in MeOH (0.9 mL) was added K2CO3 (148 mg, 1.1 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 12a (68 mg, 71%) as a colorless oil. [α] = –14.5 (c 0.29, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.38 (q, J = 2.3 Hz, 1H), 3.79 (tt, J = 8.1, 2.9 Hz, 3H), 2.38 (d, J = 2.1 Hz, 1H), 1.84–2.09 (m, 2H), 0.90 (d, J = 1.4 Hz, 18H), 0.09–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 82.4, 74.0, 73.2, 66.9, 60.1, 34.8, 25.7, 18.1, 18.0, −4.6, −4.7, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H38O3Si2Na 381.2257, found 338.2297.
(3S,4R)-3,4-bis((tert-butyldimethylsilyl)oxy)hex-5-yn-1-ol (12b): To a solution of S3b (206 mg, 0.39 mmol) in MeOH (1.3 mL) was added K2CO3 (213 mg, 1.5 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with H2O, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 30:1) to give 12b (104 mg, 76%) as a colorless oil. [α] = –22.3 (c 1.60, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.36 (q, J = 2.2 Hz, 1H), 3.94 (q, J = 4.8 Hz, 1H), 3.78 (td, J = 5.8, 2.3 Hz, 2H), 2.41 (d, J = 2.1 Hz, 1H), 1.82–2.06 (m, 2H), 0.90 (d, J = 1.7 Hz, 18H), 0.09–0.17 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 83.5, 74.1, 66.9, 59.0, 34.6, 25.9, 25.7, 18.2, 18.1, −4.3, −4.6, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H38O3Si2Na 381.2257, found 338.2263.
(5S,6S)-5-ethynyl-6-(2-iodoethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (S4a): To a solution of 12a (49 mg, 0.38 mmol) in THF (0.6 mL) was added triphenylphosphine (70 mg, 0.33 mmol), imidazole (28 mg, 0.41 mmol), and iodine (112 mg, 0.44 mmol) at –20 °C. The resulting mixture was stirred at the same temperature for 30 min. The resulting mixture was allowed to warm to room temperature and stirred for 20 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give S4a (54 mg, 83%) as a yellow oil. [α] = –35.2 (c 0.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.37 (q, J = 2.3 Hz, 1H), 3.68–3.74 (m, 1H), 3.38 (qd, J = 5.4, 3.8 Hz, 1H), 3.22 (td, J = 9.5, 6.3 Hz, 1H), 2.33 (d, J = 2.1 Hz, 1H), 2.21–2.30 (m, 1H), 2.04–2.15 (m, 1H), 0.90 (d, J = 0.7 Hz, 18H), 0.11–0.14 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 82.6, 73.9, 73.5, 66.3, 35.4, 31.1, 25.7, 18.1, 18.0, 4.2, −4.3, −4.5, −4.7, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H37IO2Si2Na 491.1274, found 491.1255.
(5R,6S)-5-ethynyl-6-(2-iodoethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (S4b): To a solution of 12b (136 mg, 0.38 mmol) in THF (1.6 mL) was added triphenylphosphine (193 mg, 0.91 mmol), imidazole (77.2 mg, 1.1 mmol), and iodine (308 mg, 1.2 mmol) at –20 °C. The resulting mixture was stirred at the same temperature for 30 min. The resulting mixture was allowed to warm to room temperature and stirred for 20 min. The reaction mixture was quenched with sat. Na2S2O3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give S4b (149 mg, 84%) as a yellow oil. [α] = –28.3 (c 1.54, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.26 (q, J = 2.1 Hz, 1H), 3.77 (td, J = 5.3, 3.3 Hz, 1H), 3.19–3.35 (m, 2H), 2.39 (d, J = 2.4 Hz, 1H), 2.11–2.20 (m, 2H), 0.90 (d, J = 2.1 Hz, 18H), 0.12–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 83.3, 75.9, 74.0, 66.8, 37.1, 25.9, 25.8, 3.1, −4.1, −4.5, −5.2; HRMS (ESI) m/z: [M + Na]+ calcd for C18H37IO2Si2Na 491.1274, found 491.1243.
(4S,5R)-4,5-bis((tert-butyldimethylsilyl)oxy)hept-6-ynenitrile (13a). To a solution of S4a (54 mg, 0.11 mmol) in DMF (0.3 mL) was added sodium cyanide (8.4 mg, 0.17 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 20 min. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 80:1) to give 13a (42 mg, 99%) as a yellow oil. [α] = –27.6 (c 0.29, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.38 (q, J = 2.3 Hz, 1H), 3.67–3.73 (m, 1H), 2.39–2.58 (m, 2H), 2.36 (d, J = 2.1 Hz, 1H), 2.04–2.16 (m, 1H), 1.90–2.02 (m, 1H), 0.90 (s, 18H), 0.11–0.14 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 119.9, 82.0, 74.0, 72.3, 66.4, 31.1, 27.6, 25.7, 18.1, 18.0, −4.6, −4.8, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H37NO2Si2Na 390.2261, found 390.2290.
(4S,5R)-4,5-bis((tert-butyldimethylsilyl)oxy)hept-6-ynenitrile (13b): To a solution of S4b (133 mg, 0.28 mmol) in DMF (0.7 mL) was added sodium cyanide (21.0 mg, 0.43 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 20 min. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 80:1) to give 13b (105 mg, 99%) as a yellow oil. [α] = –32.0 (c 2.14, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.27 (q, J = 2.1 Hz, 1H), 3.82 (td, J = 5.3, 3.2 Hz, 1H), 2.45–2.51 (m, 2H), 2.42 (d, J = 2.1 Hz, 1H), 1.89–2.12 (m, 2H), 0.90 (s, 18H), 0.11–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 120.3, 83.0, 74.4, 73.7, 66.7, 28.2, 25.8, 25.7, 18.1, 13.0, −4.2, −4.6, −4.8, −5.2; HRMS (ESI) m/z: [M + Na]+ calcd for C19H37NO2Si2Na 390.2261, found 390.2249.
(5S,6S)-5-(but-3-en-1-yl)-6-ethynyl-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (8a): To a solution of 13a (42 mg, 0.11 mmol) in CH2Cl2 (0.6 mL) was added diisobutylaluminium hydride (1.0 M in hexane; 0.14 mL, 0.14 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min. To the reaction mixture was added 2-propanol (0.098 mL), silica gel (200 mg), and water (1 mL) at room temperature. The resulting mixture was stirred at the same temperature for 30 min. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of methyltriphenylphosphonium iodide (240 mg, 0.39 mmol) in THF (0.6 mL) was added sodium bis(trimethylsilyl)amide (1.9 M in THF, 0.29 mL, 0.56 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.2 M, 0.55 mL, 0.11 mmol) was added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with n-hexane three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give 8a (27 mg, 64%) as a colorless oil. [α] = –18.0 (c 0.22, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.78–5.88 (m, 1H), 4.94–5.06 (m, 2H), 4.34 (q, J = 2.3 Hz, 1H), 3.57–3.61 (m, 1H), 2.33 (d, J = 2.3 Hz, 1H), 2.18–2.27 (m, 1H), 2.02–2.11 (m, 1H), 1.78–1.86 (m, 1H), 1.67–1.75 (m, 1H), 0.90 (s, 18H), 0.07–0.14 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 138.9, 114.3, 83.0, 74.0, 73.2, 66.8, 30.8, 29.6, 25.8, 25.8, 18.2, 18.1, −4.4, −4.5, −4.7, −5.0; HRMS (ESI) m/z: [M + Na]+ calcd for C20H40O2Si2Na 391.2465, found 391.2465.
(5S,6R)-5-(but-3-en-1-yl)-6-ethynyl-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane (8b): To a solution of 13b (32.5 mg, 0.089 mmol) in CH2Cl2 (0.4 mL) was added diisobutylaluminium hydride (1.0 M in hexane; 0.11 mL, 0.11 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min. To the reaction mixture was added 2-propanol (0.074 mL), silica gel (200 mg), and water (1 mL) at room temperature. The resulting mixture was stirred at the same temperature for 30 min. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane/ethyl acetate = 10:1) to give the aldehyde as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of methyltriphenylphosphonium iodide (189 mg, 0.47 mmol) in THF (0.5 mL) was added sodium bis(trimethylsilyl)amide (1.9 M in THF, 0.23 mL, 0.44 mmol) at 0 °C. The resulting mixture was stirred at the same temperature for 30 min, then a solution of the aldehyde in THF (0.2 M, 0.45 mL, 0.089 mmol) was added dropwise at the same temperature. The resulting mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl aq, and the aqueous layer was extracted with n-hexane three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give 8b (23 mg, 70%) as a colorless oil. [α] = –29.1 (c 3.30, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.76–5.89 (m, 1H), 4.93–5.05 (m, 2H), 4.23 (q, J = 2.3 Hz, 1H), 3.73 (q, J = 5.2 Hz, 1H), 2.35 (d, J = 2.4 Hz, 1H), 2.13 (q, J = 7.3 Hz, 2H), 1.59–1.82 (m, 2H), 0.91 (s, 18H), 0.08–0.15 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 138.8, 114.3, 84.2, 75.2, 73.3, 66.7, 32.0, 28.9, 26.0, 25.8, 18.2, −4.1, −4.5, −4.5, −5.1; HRMS (ESI) m/z: [M + Na]+ calcd for C20H40O2Si2Na 391.2465, found 391.2442.
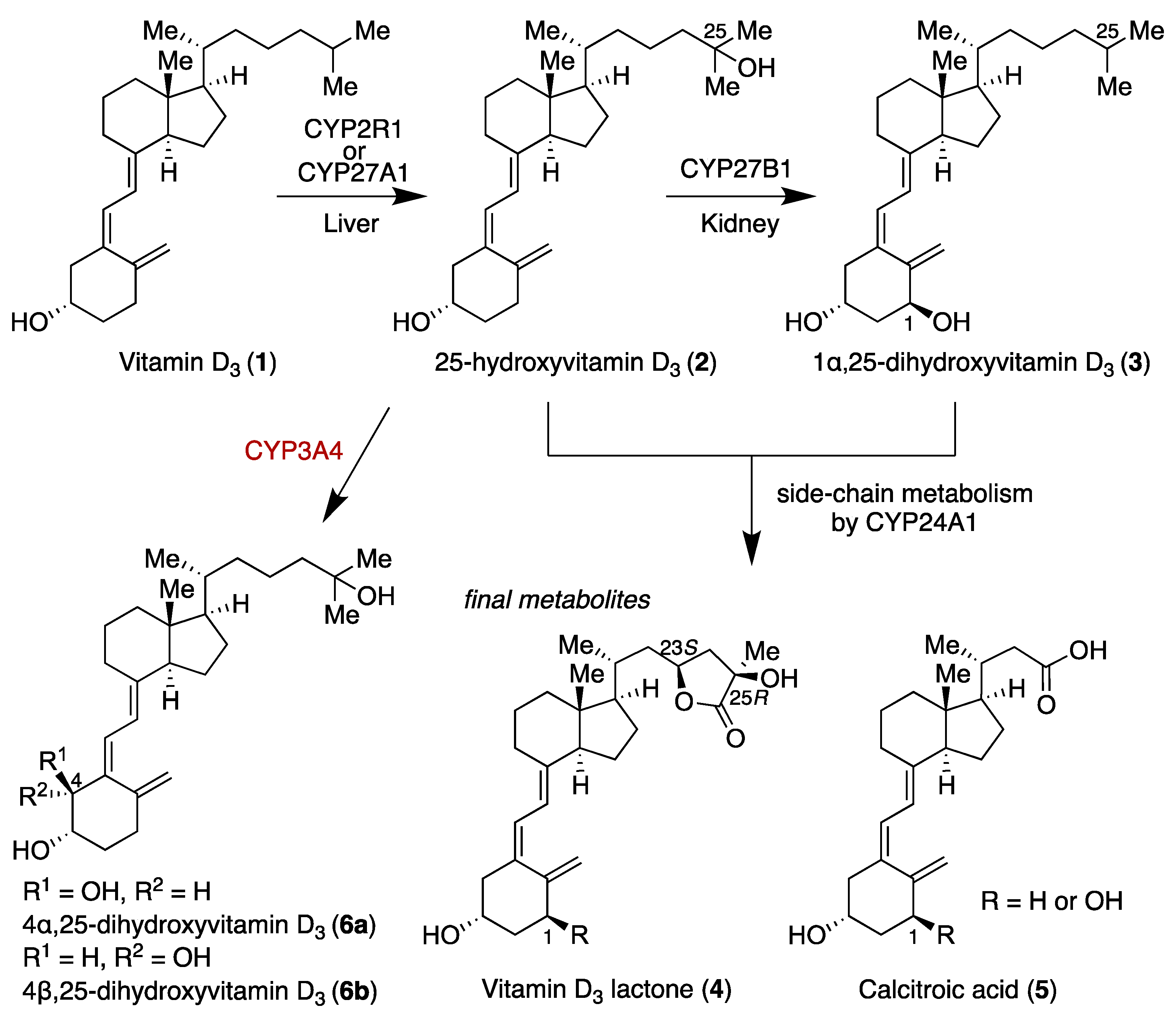
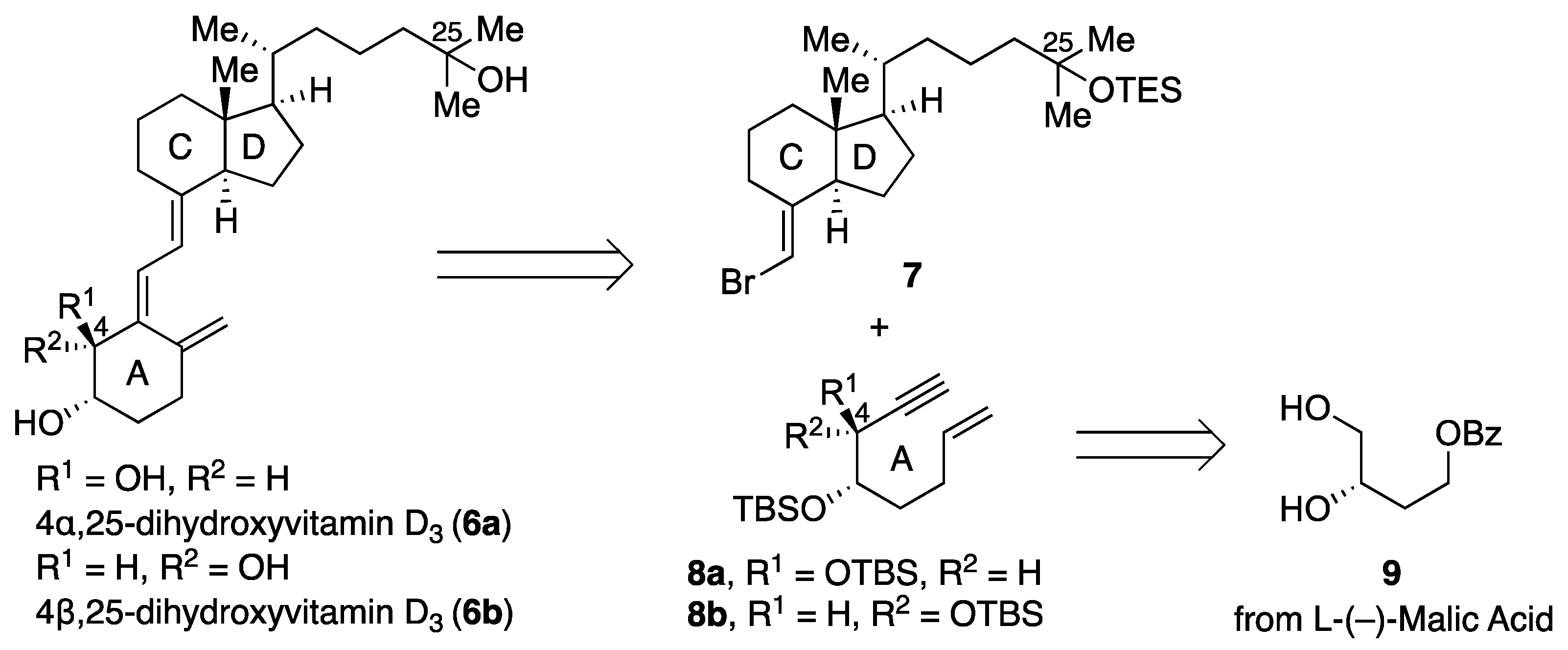
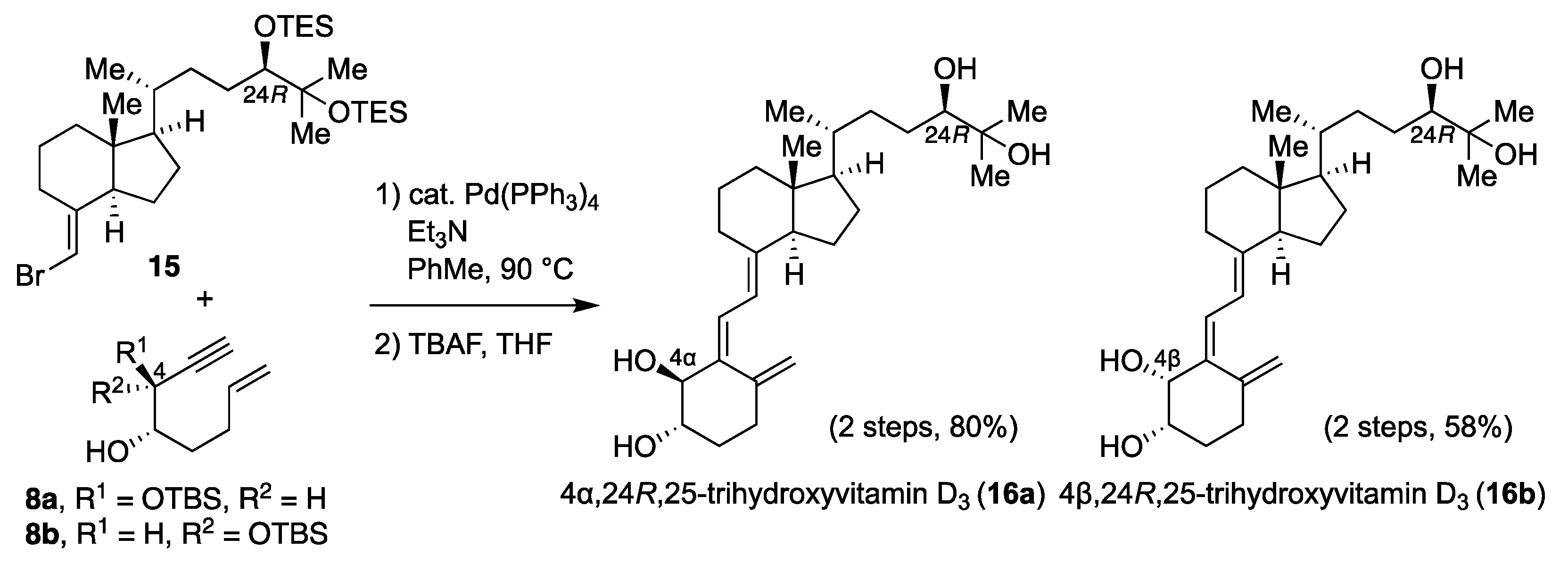
4α,25-dihydroxyvitamin D
3 (
6a): To a solution of CD-ring
7 [
14] (19 mg, 0.040 mmol),
8a (18 mg, 0.048 mmol), and triethylamine (0.4 mL) in toluene (0.4 mL) was added tetrakis(triphenylphosphine)palladium(0) (5 mg, 0.0040 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (
n-hexane) to give the coupling product (20 mg, 66%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (20 mg, 0.026 mmol) in THF (0.3 mL) was added tetra-
n-butylammonium fluoride (1.0 M in THF, 0.26 mL, 0.26 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO
3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO
4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:20) to give
6a (8 mg, 77%) as a yellow oil. [α]
= +162.5 (
c 0.08, CHCl
3);
1H NMR (400 MHz, CDCl
3) δ 6.58 (d,
J = 10.1 Hz, 1H), 6.04 (d,
J = 11.4 Hz, 1H), 5.12 (s, 1H), 4.87 (s, 1H), 3.96 (d,
J = 8.2 Hz, 1H), 3.57 (td,
J = 8.9, 3.7 Hz, 1H), 2.90 (d,
J = 12.8 Hz, 1H), 2.36 (td,
J = 8.8, 4.4 Hz, 1H), 2.16–2.21 (m, 1H), 2.07–2.13 (m, 1H), 2.00 (t,
J = 9.2 Hz, 2H), 1.88 (t,
J = 8.2 Hz, 1H), 1.21–1.82 (m, 22H), 0.93 (d,
J = 6.4 Hz, 3H), 0.53 (s, 3H);
13C NMR (100 MHz, CDCl
3) δ 144.1, 143.5, 137.3, 119.1, 117.1, 114.6, 78.4, 74.6, 71.1, 56.5, 56.4, 52.1, 45.9, 44.4, 40.5, 36.4, 36.1, 32.2, 29.7, 29.4, 29.2, 27.7, 23.5, 22.2, 20.8, 18.8, 12.0; HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
27H
44O
3Na 439.3188, found 439.3177.
4β,25-dihydroxyvitamin D3 (6b): To a solution of CD-ring 7 (38 mg, 0.080 mmol), 8b (35 mg, 0.096 mmol), and triethylamine (0.8 mL) in toluene (0.8 mL) was added tetrakis(triphenylphosphine)palladium(0) (9 mg, 0.0080 mmol) at room temperature. The resulting mixture was allowed to warm to 90 °C and stirred for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (n-hexane) to give the coupling product (39 mg, 65%) as a yellow oil. The crude residue was used in the subsequent step without purification. To a solution of the coupling product (9 mg, 0.012 mmol) in THF (0.6 mL) was added tetra-n-butylammonium fluoride (1.0 M in THF, 0.12 mL, 0.12 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 12 h. The reaction mixture was quenched with sat. NaHCO3 aq, and the aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (methanol/chloroform 1:20) to give 6b (5 mg, 100%) as a yellow oil. [α] = +39.0 (c 0.21, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.51 (d, J = 10.9 Hz, 1H), 6.04 (d, J = 11.5 Hz, 1H), 5.17 (s, 1H), 4.92 (s, 1H), 4.22 (s, 1H), 3.86 (s, 1H), 2.86 (d, J = 13.2 Hz, 1H), 2.35–2.38 (m, 1H), 2.13–2.18 (m, 1H), 2.00–2.02 (m, 2H), 1.84–1.85 (m, 3H), 1.21–1.71 (m, 21H), 0.93 (d, J = 6.3 Hz, 3H), 0.54 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 145.4, 142.6, 137.2, 123.5, 117.0, 115.4, 71.9, 71.1, 56.5, 56.4, 46.0, 44.4, 40.5, 36.4, 36.1, 32.0, 30.1, 29.7, 29.4, 29.2, 29.2, 27.7, 23.6, 22.2, 20.8, 18.8, 12.0; HRMS (ESI) m/z: [M + Na]+ calcd for C27H44O3Na 439.3188, found 439.3181.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}