
How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Review of the Literature
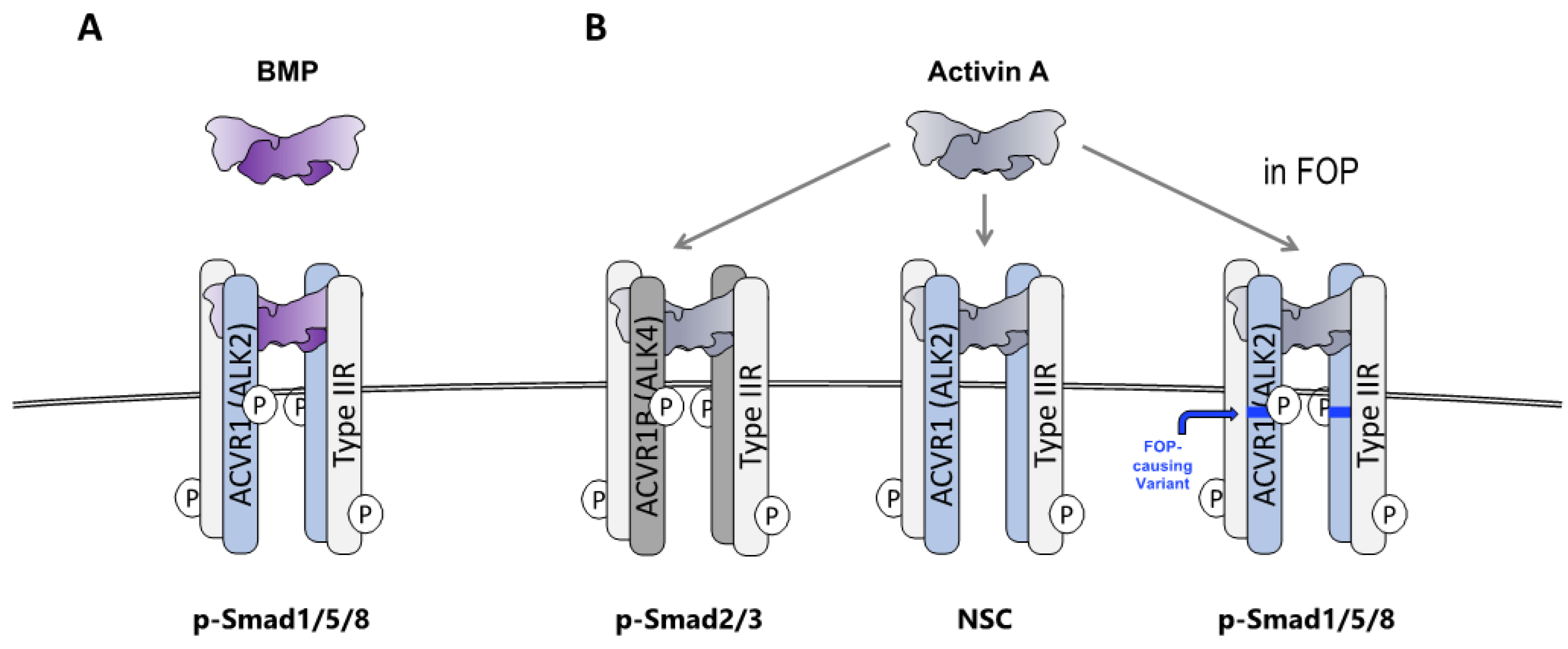
2.1. HO in FOP Requires Activation of ACVR1FOP by Ligand
2.2. The Ligand That Drives HO in FOP Is Activin A
2.3. FOP Variant ACVR1 Is Activated by Activin Whereas Wild Type ACVR1 Is Not
2.4. Inhibition of Activin A Blocks HO in Patients with FOP
2.5. Anti-ACVR1 Antibodies Activate Signaling by FOP-Variant ACVR1
2.6. Fibroadipogenic Progenitors Are the Heterotopic Bone-Forming Cells in FOP
2.7. Wild-Type ACVR1 Forms Non-Signaling Complexes with Activin A and Cognate Type II Receptors
2.8. The ACVR1•Activin A•Type II Receptor Non-Signaling Complex Tempers the Severity of HO in FOP Mice
3. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, R.B.; Hahn, G.V.; Tabas, J.A.; Peeper, J.; Levitz, C.L.; Sando, A.; Sando, N.; Zasloff, M.; Kaplan, F.S. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J. Bone Jt. Surg. Am. 1993, 75, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Huning, I.; Gillessen-Kaesbach, G. Fibrodysplasia ossificans progressiva: Clinical course, genetic mutations and genotype-phenotype correlation. Mol. Syndromol. 2014, 5, 201–211. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Baujat, G.; Brown, M.A.; De Cunto, C.; Di Rocco, M.; Hsiao, E.C.; Keen, R.; Al Mukaddam, M.; Sang, K.L.Q.; Wilson, A.; et al. Natural history of fibrodysplasia ossificans progressiva: Cross-sectional analysis of annotated baseline phenotypes. Orphanet J. Rare Dis. 2019, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J.; Brahim, J.S.; Zasloff, M.A.; Marini, J.C. Failure of surgery and isotretinoin to relieve jaw immobilization in fibrodysplasia ossificans progressiva: Report of two cases. J. Oral. Maxillofac. Surg. 1990, 48, 204–208. [Google Scholar] [CrossRef]
- Botman, E.; Treurniet, S.; Lubbers, W.D.; Schwarte, L.A.; Schober, P.R.; Sabelis, L.; Peters, E.J.G.; van Schie, A.; de Vries, R.; Grunwald, Z.; et al. When Limb Surgery Has Become the Only Life-Saving Therapy in FOP: A Case Report and Systematic Review of the Literature. Front. Endocrinol. 2020, 11, 570. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthrom, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Baujat, G.; Brown, M.A.; De Cunto, C.; Hsiao, E.C.; Keen, R.; Al Mukaddam, M.; Le Quan Sang, K.H.; Wilson, A.; Marino, R.; et al. The natural history of fibrodysplasia ossificans progressiva: A prospective, global 36-month study. Genet. Med. 2022, 24, 2422–2433. [Google Scholar] [CrossRef]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Heterotopic bone induction via BMP signaling: Potential therapeutic targets for fibrodysplasia ossificans progressiva. Bone 2018, 109, 241–250. [Google Scholar] [CrossRef]
- Shafritz, A.B.; Shore, E.M.; Gannon, F.H.; Zasloff, M.A.; Taub, R.; Muenke, M.; Kaplan, F.S. Overexpression of an Osteogenic Morphogen in Fibrodysplasia Ossificans Progressiva. N. Engl. J. Med. 1996, 335, 555–561. [Google Scholar] [CrossRef]
- Olmsted, E.A.; Kaplan, F.S.; Shore, E.M. Bone morphogenetic protein-4 regulation in fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 2003, 408, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Hu, M.; Gomes, W.A.; Kessler, J.A. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am. J. Pathol. 2004, 165, 1107–1115. [Google Scholar] [CrossRef]
- Glaser, D.L.; Economides, A.N.; Wang, L.; Liu, X.; Kimble, R.D.; Fandl, J.P.; Wilson, J.M.; Stahl, N.; Kaplan, F.S.; Shore, E.M. In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4-induced heterotopic ossification. J. Bone Jt. Surg. Am. 2003, 85, 2332–2342. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Suda, R.K.; Kaplan, F.S. The fibrodysplasia ossificans progressiva lesion. Clin. Rev. Bone Miner. Metab. 2005, 3, 195–200. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Rigueur, D.; Brugger, S.; Anbarchian, T.; Kim, J.K.; Lee, Y.; Lyons, K.M. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 2015, 30, 733–741. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef]
- Wieser, R.; Wrana, J.; Massague, J. GS domain mutations that constitutively activate T beta RI, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995, 14, 2199. [Google Scholar] [CrossRef]
- Fukuda, T.; Scott, G.; Komatsu, Y.; Araya, R.; Kawano, M.; Ray, M.K.; Yamada, M.; Mishina, Y. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 2006, 44, 159–167. [Google Scholar] [CrossRef]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef]
- Dinther, M.v.; Visser, N.; de Gorter, D.J.J.; Doorn, J.; Goumans, M.-J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia Ossificans Progressiva: Clinical and Genetic Aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Smilde, B.J.; Botman, E.; de Ruiter, R.D.; Smit, J.M.; Teunissen, B.P.; Lubbers, W.D.; Schwarte, L.A.; Schober, P.; Eekhoff, E.M.W. Monitoring and Management of Fibrodysplasia Ossificans Progressiva: Current Perspectives. Orthop. Res. Rev. 2022, 14, 113–120. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Bartnikas, T.B.; Lohmeyer, L.K.; Leyton, P.; Mayeur, C.; Kao, S.M.; Pappas, A.E.; Peterson, R.T.; Bloch, D.B.; Yu, P.B.; et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 2011, 118, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Pagani, A.; Colucci, S.; Bocciardi, R.; Bertamino, M.; Dufour, C.; Ravazzolo, R.; Silvestri, L.; Camaschella, C. A new form of IRIDA due to combined heterozygous mutations of TMPRSS6 and ACVR1A encoding the BMP receptor ALK2. Blood 2017, 129, 3392–3395. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Vanhoutte, F.; Liang, S.; Ruddy, M.; Zhao, A.; Drewery, T.; Wang, Y.; DelGizzi, R.; Forleo-Neto, E.; Rajadhyaksha, M.; Herman, G.; et al. Pharmacokinetics and Pharmacodynamics of Garetosmab (Anti-Activin A): Results From a First-in-Human Phase 1 Study. J. Clin. Pharmacol. 2020, 60, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Forleo-Neto, E.; Pignolo, R.J.; Keen, R.; Orcel, P.; Funck-Brentano, T.; Roux, C.; Kolta, S.; Madeo, A.; Bubbear, J.S.; et al. Garetosmab in fibrodysplasia ossificans progressiva: A randomized, double-blind, placebo-controlled phase 2 trial. Nat. Med. 2023, 29, 2615–2624. [Google Scholar] [CrossRef]
- Cappato, S.; Giacopelli, F.; Ravazzolo, R.; Bocciardi, R. The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva. Int. J. Mol. Sci. 2018, 19, 989. [Google Scholar] [CrossRef]
- Kitoh, H. Clinical Aspects and Current Therapeutic Approaches for FOP. Biomedicines 2020, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef]
- Schnutgen, F.; Doerflinger, N.; Calleja, C.; Wendling, O.; Chambon, P.; Ghyselinck, N.B. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 2003, 21, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, J.; Xie, L.; Huang, L.; Das, N.; Stewart, R.C.; Lyon, M.C.; Palmer, K.; Rajamani, S.; Graul, C.; Lobo, M.; et al. The Expansion of Heterotopic Bone in Fibrodysplasia Ossificans Progressiva Is Activin A-Dependent. J. Bone Miner. Res. 2017, 32, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Sako, D.; Grinberg, A.V.; Liu, J.; Davies, M.V.; Castonguay, R.; Maniatis, S.; Andreucci, A.J.; Pobre, E.G.; Tomkinson, K.N.; Monnell, T.E.; et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 2010, 285, 21037–21048. [Google Scholar] [CrossRef]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-beta family action. Cytokine Growth Factor. Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef]
- de Kretser, D.M.; O’Hehir, R.E.; Hardy, C.L.; Hedger, M.P. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell Endocrinol. 2012, 359, 101–106. [Google Scholar] [CrossRef]
- Attisano, L.; Cárcamo, J.; Ventura, F.; Weis, F.M.B.; Massagué, J.; Wrana, J.L. Identification of human activin and TGFβ type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75, 671–680. [Google Scholar] [CrossRef]
- Ebner, R.; Chen, R.H.; Lawler, S.; Zioncheck, T.; Derynck, R. Determination of type I receptor specificity by the type II receptors for TGF-beta or activin. Science 1993, 262, 900–902. [Google Scholar] [CrossRef]
- Tsuchida, K.; Mathews, L.S.; Vale, W.W. Cloning and characterization of a transmembrane serine kinase that acts as an activin type I receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11242–11246. [Google Scholar] [CrossRef]
- Latres, E.; Mastaitis, J.; Fury, W.; Miloscio, L.; Trejos, J.; Pangilinan, J.; Okamoto, H.; Cavino, K.; Na, E.; Papatheodorou, A.; et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 15153. [Google Scholar] [CrossRef]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, e54582. [Google Scholar] [CrossRef]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef]
- Cappato, S.; Traberg, R.; Gintautiene, J.; Zara, F.; Bocciardi, R. A case of Fibrodysplasia Ossificans Progressiva associated with a novel variant of the ACVR1 gene. Mol. Genet. Genom. Med. 2021, 9, e1774. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M.; Tsuji, S.; Nakamura, K.; Ohte, S.; Kawaguchi, Y.; Takaishi, K. A blocking monoclonal antibody reveals dimerization of intracellular domains of ALK2 associated with genetic disorders. Nat. Commun. 2023, 14, 2960. [Google Scholar] [CrossRef]
- Aykul, S.; Huang, L.; Wang, L.; Das, N.M.; Reisman, S.; Ray, Y.; Zhang, Q.; Rothman, N.; Nannuru, K.C.; Kamat, V.; et al. Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1. J. Clin. Investig. 2022, 132, e153792. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Stoessel, S.J.; Chandler, J.T.; Bouchard, K.; Bento, P.; Apuzzo, L.N.; Devarakonda, P.M.; Hunter, J.W.; Goldhamer, D.J. An anti-ACVR1 antibody exacerbates heterotopic ossification by fibro-adipogenic progenitors in fibrodysplasia ossificans progressiva mice. J. Clin. Investig. 2022, 132, e153795. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Bagarova, J.; Vonner, A.J.; Armstrong, K.A.; Borgermann, J.; Lai, C.S.; Deng, D.Y.; Beppu, H.; Alfano, I.; Filippakopoulos, P.; Morrell, N.W.; et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell Biol. 2013, 33, 2413–2424. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Gilboa, L.; Nohe, A.; Geissendorfer, T.; Sebald, W.; Henis, Y.I.; Knaus, P. Bone morphogenetic protein receptor complexes on the surface of live cells: A new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell 2000, 11, 1023–1035. [Google Scholar] [CrossRef]
- Zhou, Y.; Scolavino, S.; Funderburk, S.F.; Ficociello, L.F.; Zhang, X.; Klibanski, A. Receptor internalization-independent activation of Smad2 in activin signaling. Mol. Endocrinol. 2004, 18, 1818–1826. [Google Scholar] [CrossRef]
- Ehrlich, M.; Horbelt, D.; Marom, B.; Knaus, P.; Henis, Y.I. Homomeric and heteromeric complexes among TGF-beta and BMP receptors and their roles in signaling. Cell Signal 2011, 23, 1424–1432. [Google Scholar] [CrossRef]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Pryce, B.A.; Brent, A.E.; Murchison, N.D.; Tabin, C.J.; Schweitzer, R. Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Dev. Dyn. 2007, 236, 1677–1682. [Google Scholar] [CrossRef]
- Park, D.; Spencer, J.A.; Koh, B.I.; Kobayashi, T.; Fujisaki, J.; Clemens, T.L.; Lin, C.P.; Kronenberg, H.M.; Scadden, D.T. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012, 10, 259–272. [Google Scholar] [CrossRef]
- Scott, R.W.; Arostegui, M.; Schweitzer, R.; Rossi, F.M.V.; Underhill, T.M. Hic1 Defines Quiescent Mesenchymal Progenitor Subpopulations with Distinct Functions and Fates in Skeletal Muscle Regeneration. Cell Stem Cell 2019, 25, 797–813 e799. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef]
- Oishi, T.; Uezumi, A.; Kanaji, A.; Yamamoto, N.; Yamaguchi, A.; Yamada, H.; Tsuchida, K. Osteogenic differentiation capacity of human skeletal muscle-derived progenitor cells. PLoS ONE 2013, 8, e56641. [Google Scholar] [CrossRef]
- Penton, C.M.; Thomas-Ahner, J.M.; Johnson, E.K.; McAllister, C.; Montanaro, F. Muscle side population cells from dystrophic or injured muscle adopt a fibro-adipogenic fate. PLoS ONE 2013, 8, e54553. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef]
- Eisner, C.; Cummings, M.; Johnston, G.; Tung, L.W.; Groppa, E.; Chang, C.; Rossi, F.M. Murine Tissue-Resident PDGFRalpha+ Fibro-Adipogenic Progenitors Spontaneously Acquire Osteogenic Phenotype in an Altered Inflammatory Environment. J. Bone Miner. Res. 2020, 35, 1525–1534. [Google Scholar] [CrossRef]
- Lemos, D.R.; Paylor, B.; Chang, C.; Sampaio, A.; Underhill, T.M.; Rossi, F.M. Functionally convergent white adipogenic progenitors of different lineages participate in a diffused system supporting tissue regeneration. Stem Cells 2012, 30, 1152–1162. [Google Scholar] [CrossRef]
- Harvey, T.; Flamenco, S.; Fan, C.M. A Tppp3+Pdgfra+ tendon stem cell population contributes to regeneration and reveals a shared role for PDGF signalling in regeneration and fibrosis. Nat. Cell Biol. 2019, 21, 1490–1503. [Google Scholar] [CrossRef]
- Armes, N.A.; Smith, J.C. The ALK-2 and ALK-4 activin receptors transduce distinct mesoderm-inducing signals during early Xenopus development but do not co-operate to establish thresholds. Development 1997, 124, 3797–3804. [Google Scholar] [CrossRef]
- Chen, Y.; Bhushan, A.; Vale, W. Smad8 mediates the signaling of the ALK-2 [corrected] receptor serine kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 12938–12943. [Google Scholar] [CrossRef]
- Macías-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific Activation of Smad1 Signaling Pathways by the BMP7 Type I Receptor, ALK2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef]
- Piek, E.; Afrakhte, M.; Sampath, K.; van Zoelen, E.J.; Heldin, C.H.; ten Dijke, P. Functional antagonism between activin and osteogenic protein-1 in human embryonal carcinoma cells. J. Cell Physiol. 1999, 180, 141–149. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef]
- Goebel, E.J.; Corpina, R.A.; Hinck, C.S.; Czepnik, M.; Castonguay, R.; Grenha, R.; Boisvert, A.; Miklossy, G.; Fullerton, P.T.; Matzuk, M.M.; et al. Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 15505–15513. [Google Scholar] [CrossRef]
- Yamamoto, M.; Stoessel, S.J.; Yamamoto, S.; Goldhamer, D.J. Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification. J. Bone Miner. Res. 2022, 37, 2077–2093. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders. Biomedicines 2021, 9, 736. [Google Scholar] [CrossRef]
- Ventura, F.; Williams, E.; Ikeya, M.; Bullock, A.N.; Ten Dijke, P.; Goumans, M.J.; Sanchez-Duffhues, G. Challenges and Opportunities for Drug Repositioning in Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 213. [Google Scholar] [CrossRef]
- Valer, J.A.; Sanchez-de-Diego, C.; Pimenta-Lopes, C.; Rosa, J.L.; Ventura, F. ACVR1 Function in Health and Disease. Cells 2019, 8, 1366. [Google Scholar] [CrossRef]
- Lin, H.; Shi, F.; Gao, J.; Hua, P. The role of Activin A in fibrodysplasia ossificans progressiva: A prominent mediator. Biosci. Rep. 2019, 39, BSR20190377. [Google Scholar] [CrossRef]
- Wentworth, K.L.; Masharani, U.; Hsiao, E.C. Therapeutic advances for blocking heterotopic ossification in fibrodysplasia ossificans progressiva. Br. J. Clin. Pharmacol. 2019, 85, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Tsukamoto, S.; Nakachi, Y.; Kuratani, M. Recent Topics in Fibrodysplasia Ossificans Progressiva. Endocrinol. Metab. 2018, 33, 331–338. [Google Scholar] [CrossRef]
- Wang, H.; Shore, E.M.; Pignolo, R.J.; Kaplan, F.S. Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, W.; Lacroix, J.J.; Luo, Y.; Hao, J. Insight into Molecular Mechanism for Activin A-Induced Bone Morphogenetic Protein Signaling. Int. J. Mol. Sci. 2020, 21, 6498. [Google Scholar] [CrossRef]
- Ramachandran, A.; Mehic, M.; Wasim, L.; Malinova, D.; Gori, I.; Blaszczyk, B.K.; Carvalho, D.M.; Shore, E.M.; Jones, C.; Hyvonen, M.; et al. Pathogenic ACVR1(R206H) activation by Activin A-induced receptor clustering and autophosphorylation. EMBO J. 2021, 40, e106317. [Google Scholar] [CrossRef]
- Mundy, C.; Yao, L.; Shaughnessy, K.A.; Saunders, C.; Shore, E.M.; Koyama, E.; Pacifici, M. Palovarotene Action Against Heterotopic Ossification Includes a Reduction of Local Participating Activin A-Expressing Cell Populations. JBMR Plus 2023, 7, e10821. [Google Scholar] [CrossRef]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002, 277, 5330–5338. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srinivasan, D.; Arostegui, M.; Goebel, E.J.; Hart, K.N.; Aykul, S.; Lees-Shepard, J.B.; Idone, V.; Hatsell, S.J.; Economides, A.N. How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva. Biomolecules 2024, 14, 101. https://doi.org/10.3390/biom14010101
Srinivasan D, Arostegui M, Goebel EJ, Hart KN, Aykul S, Lees-Shepard JB, Idone V, Hatsell SJ, Economides AN. How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva. Biomolecules. 2024; 14(1):101. https://doi.org/10.3390/biom14010101
Chicago/Turabian StyleSrinivasan, Dushyanth, Martin Arostegui, Erich J. Goebel, Kaitlin N. Hart, Senem Aykul, John B. Lees-Shepard, Vincent Idone, Sarah J. Hatsell, and Aris N. Economides. 2024. "How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva" Biomolecules 14, no. 1: 101. https://doi.org/10.3390/biom14010101
APA StyleSrinivasan, D., Arostegui, M., Goebel, E. J., Hart, K. N., Aykul, S., Lees-Shepard, J. B., Idone, V., Hatsell, S. J., & Economides, A. N. (2024). How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva. Biomolecules, 14(1), 101. https://doi.org/10.3390/biom14010101