

An Anti-CD7 Antibody–Drug Conjugate Target Showing Potent Antitumor Activity for T-Lymphoblastic Leukemia (T-ALL)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Lines and Culture Conditions
2.3. Expression and Purification of Recombinant Proteins
2.4. Generation of Monoclonal Antibody
2.5. Preparation of CD7-Dxd ADCs
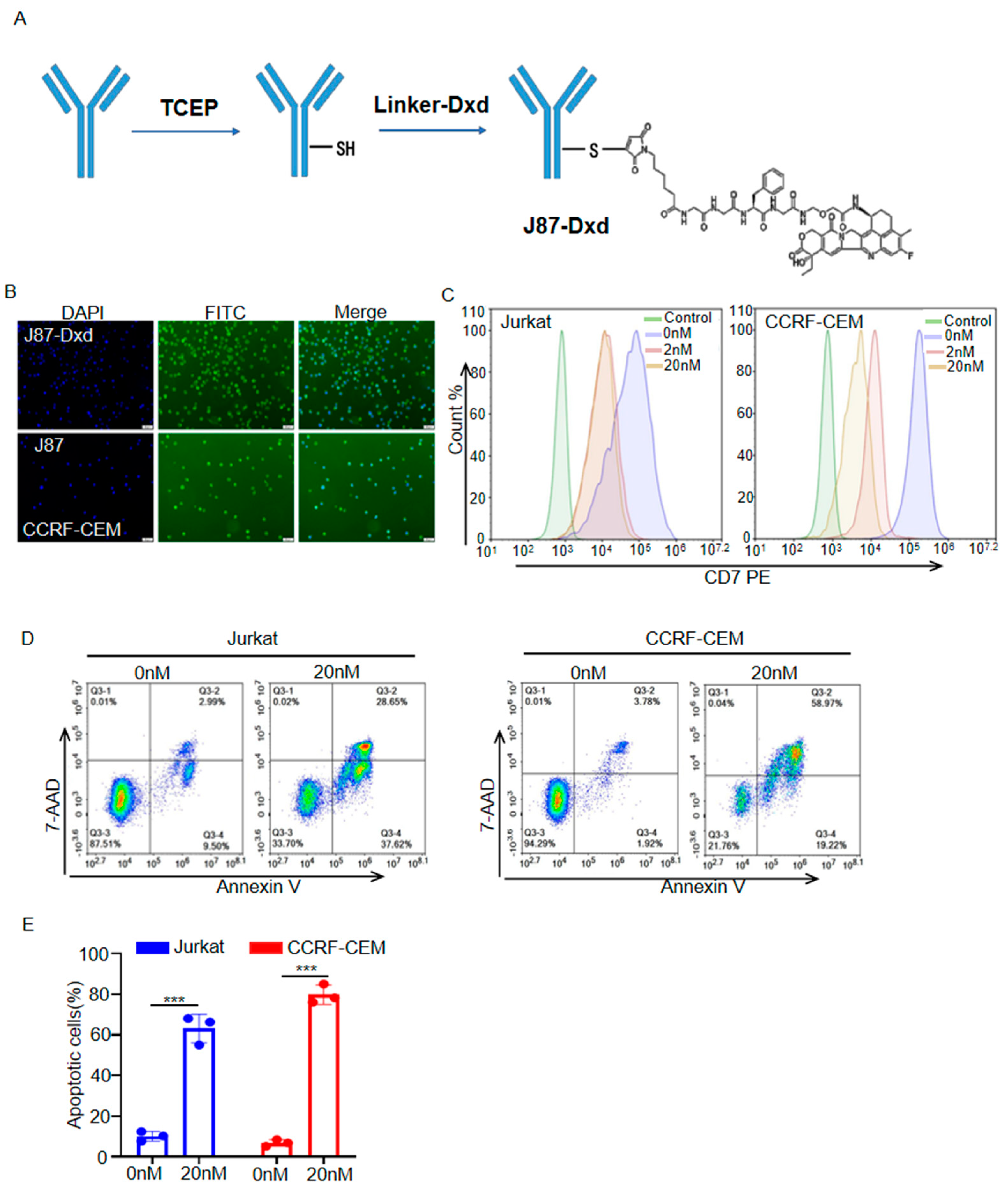
2.6. Cell Apoptosis Assay
2.7. Viability Assays
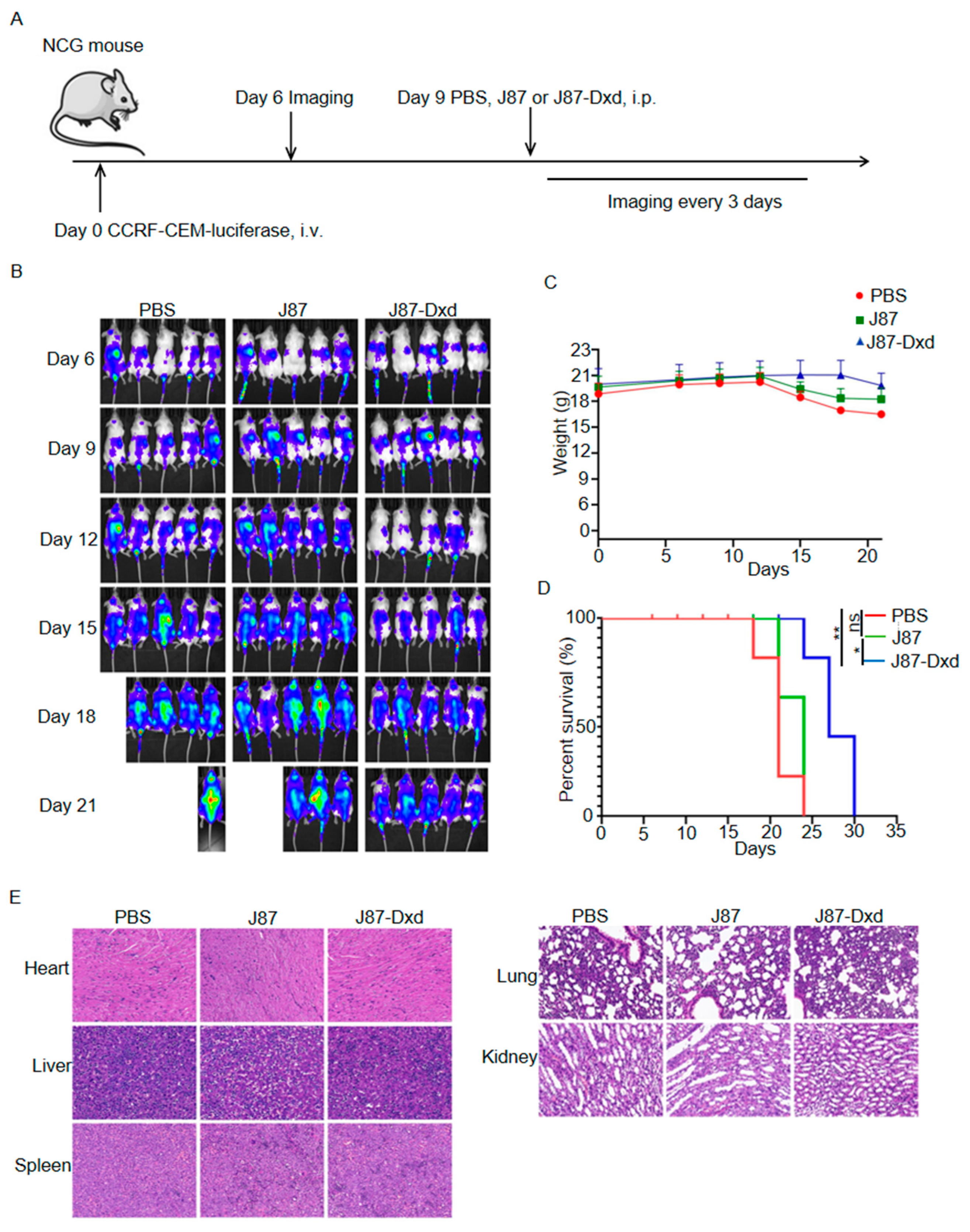
2.8. In Vivo Antitumor Analysis
2.9. Statistical Analysis
3. Results
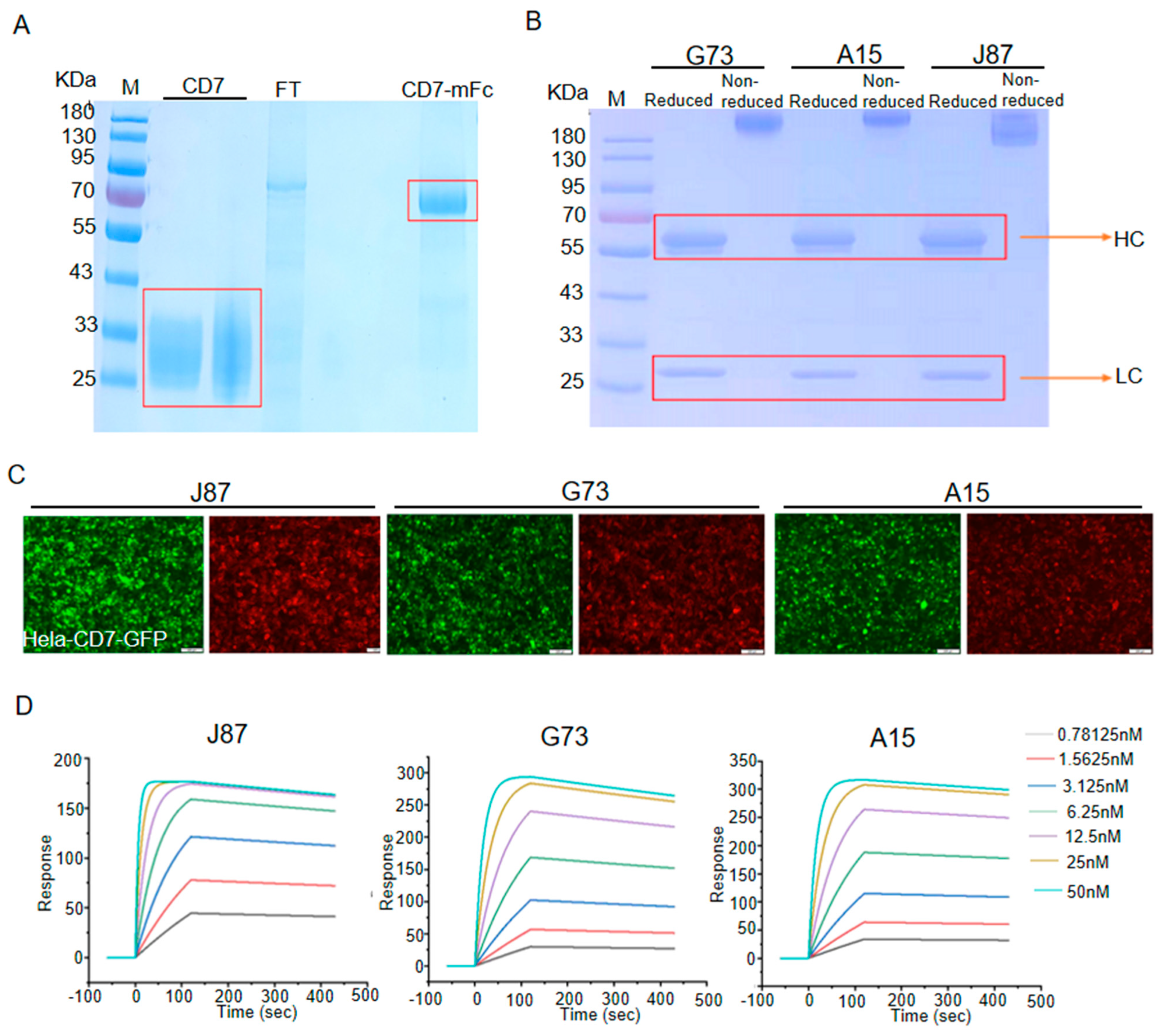
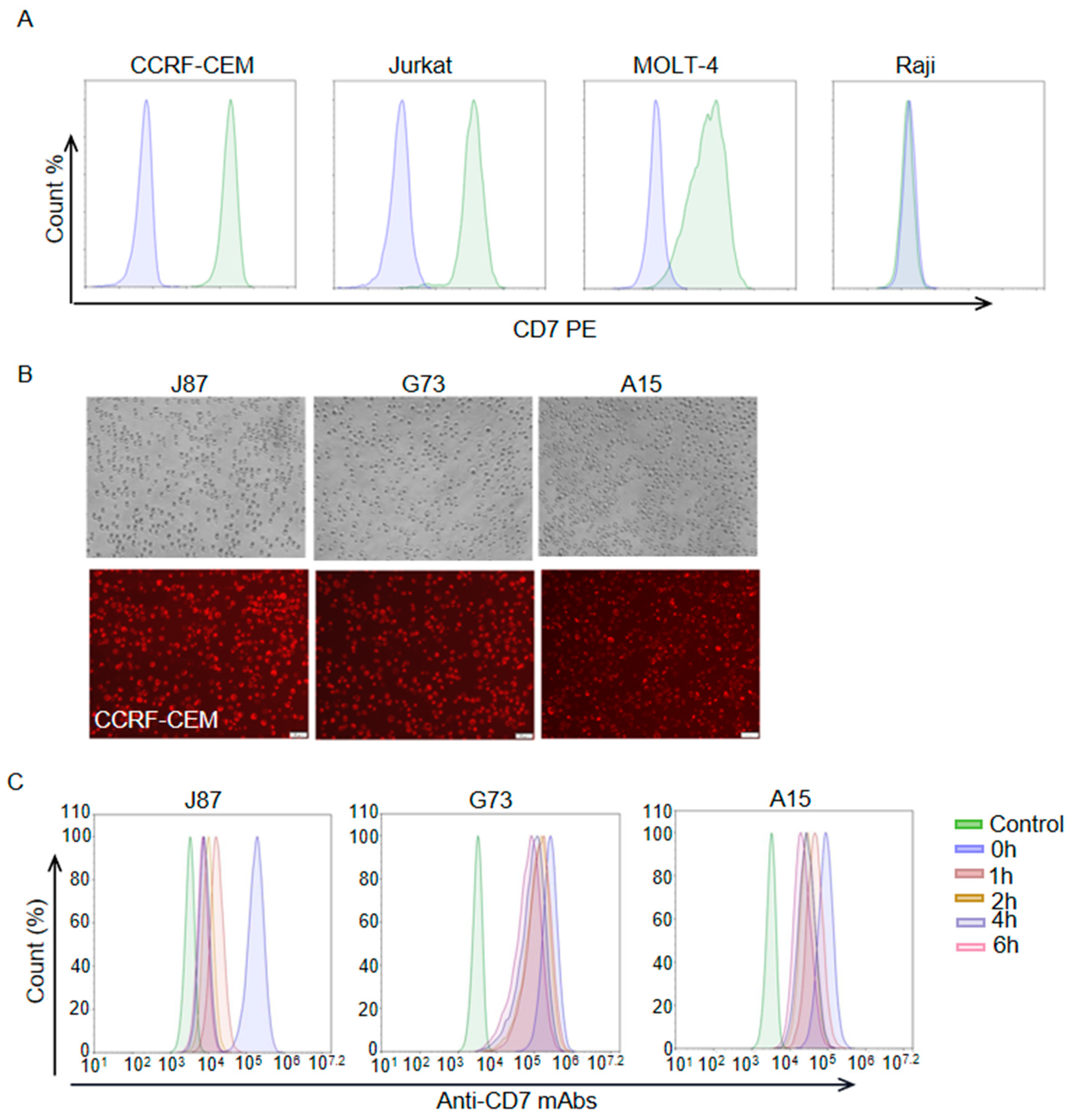
3.1. Generation and Characterization of mAbs against CD7
3.2. Internalization Assay of Anti-CD7 mAbs
3.3. Generation and Cytotoxicity Assessment of J87-Dxd
3.4. In Vivo Antitumor Efficacy of J87-Dxd in T-ALL Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef]
- Shiraz, P.; Jehangir, W.; Agrawal, V. T-Cell Acute Lymphoblastic Leukemia-Current Concepts in Molecular Biology and Management. Biomedicines 2021, 9, 1621. [Google Scholar]
- Pocock, R.; Farah, N.; Richardson, S.E.; Mansour, M.R. Current and emerging therapeutic approaches for T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2021, 194, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Zhou, S.H.; Higley, H.; Mukundan, L.; Fu, S.S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease with Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia AMeta-analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V. Relapsed ALL: CAR T vs transplant vs novel therapies. Hematol.-Am. Soc. Hematol. 2021, 2021, 1–6. [Google Scholar] [CrossRef]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody-drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 255. [Google Scholar] [PubMed]
- Kesireddy, M.; Kothapalli, S.R.; Gundepalli, S.G.; Asif, S. A Review of the Current FDA-Approved Antibody-Drug Conjugates: Landmark Clinical Trials and Indications. Pharm. Med. 2023; ahead of print. [Google Scholar]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody-Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar]
- Shah, N.N.; Sokol, L. Targeting CD22 for the Treatment of B-Cell Malignancies. ImmunoTargets Ther. 2021, 10, 225–236. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Recent updates for antibody therapy for acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2020, 9, 33. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Salma, F.; Yi, Q.L.; Patterson, B.; Brien, B.; Minden, M.D. Prognostic relevance of immunophenotyping in 379 patients with acute myeloid leukemia. Leuk. Res. 2004, 28, 43–48. [Google Scholar] [CrossRef]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Yang, D.; Chen, X.; Liang, D.; Zou, L.; Zhao, X. Chimeric antigen receptor T-cell therapy for T-ALL and AML. Front. Oncol. 2022, 12, 967754. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J.; et al. Antibody-Drug Conjugates: The New Frontier of Chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef]
- Pope, M.E.; Soste, M.V.; Eyford, B.A.; Anderson, N.L.; Pearson, T.W. Anti-peptide antibody screening: Selection of high affinity monoclonal reagents by a refined surface plasmon resonance technique. J. Immunol. Methods 2009, 341, 86–96. [Google Scholar] [CrossRef]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef]
- Ren, A.; Tong, X.; Xu, N.; Zhang, T.; Zhou, F.; Zhu, H. CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities. Vaccines 2023, 11, 165. [Google Scholar] [CrossRef]
- Zhao, W.L. Targeted therapy in T-cell malignancies: Dysregulation of the cellular signaling pathways. Leukemia 2010, 24, 13–21. [Google Scholar] [CrossRef]
- Marks, D.I.; Paietta, E.M.; Moorman, A.V.; Richards, S.M.; Buck, G.; DeWald, G.; Ferrando, A.; Fielding, A.K.; Goldstone, A.H.; Ketterling, R.P.; et al. T-cell acute lymphoblastic leukemia in adults: Clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 2009, 114, 5136–5145. [Google Scholar] [CrossRef]
- Mohty, B.; Mohty, M. Long-term complications and side effects after allogeneic hematopoietic stem cell transplantation: An update. Blood Cancer J. 2011, 1, e16. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, H.H. Targeted treatment of T-cell acute lymphoblastic leukemia: Latest updates from the 2022 ASH Annual Meeting. Exp. Hematol. Oncol. 2023, 12, 30. [Google Scholar] [CrossRef]
- Zhang, J.; Jain, A.; Milhas, S.; Williamson, D.J.; Mysliwy, J.; Lodge, A.; Thirlway, J.; Al Nakeeb, M.; Miller, A.; Rabbitts, T.H. An antibody-drug conjugate with intracellular drug release properties showing specific cytotoxicity against CD7-positive cells. Leuk. Res. 2021, 108, 106626. [Google Scholar] [CrossRef] [PubMed]
- Baum, W.; Steininger, H.; Bair, H.J.; Becker, W.; Hansen-Hagge, T.E.; Kressel, M.; Kremmer, E.; Kalden, J.R.; Gramatzki, M. Therapy with CD7 monoclonal antibody TH-69 is highly effective for xenografted human T-cell ALL. Br. J. Haematol. 1996, 95, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Peipp, M.; Küpers, H.; Saul, D.; Schlierf, B.; Greil, J.; Zunino, S.J.; Gramatzki, M.; Fey, G.H. A recombinant CD7-specific single-chain immunotoxin is a potent inducer of apoptosis in acute leukemic T cells. Cancer Res. 2002, 62, 2848–2855. [Google Scholar]
- Tang, J.; Li, J.; Zhu, X.; Yu, Y.; Chen, D.; Yuan, L.; Gu, Z.; Zhang, X.; Qi, L.; Gong, Z.; et al. Novel CD7-specific nanobody-based immunotoxins potently enhanced apoptosis of CD7-positive malignant cells. Oncotarget 2016, 7, 34070–34083. [Google Scholar] [CrossRef]
- Xie, L.; Gu, R.; Yang, X.; Qiu, S.; Xu, Y.; Mou, J.; Wang, Y.; Xing, H.; Tang, K.; Tian, Z.; et al. Universal Anti-CD7 CAR-T Cells Targeting T-ALL and Functional Analysis of CD7 Antigen on T/CAR-T Cells. Hum. Gene Ther. 2023, 34, 1257–1272. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Liu, L.; Liu, J.; Rao, J.; Yuan, Z.; Gao, L.; Li, Y.; Luo, L.; Li, G.; et al. CD7 targeted “off-the-shelf” CAR-T demonstrates robust in vivo expansion and high efficacy in the treatment of patients with relapsed and refractory T cell malignancies. Leukemia 2023, 37, 2176–2186. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAbs | Ka (1/Ms) | Kd (1/s) | KD (M) |
|---|---|---|---|
| J87 | 1.21 × 106 | 1.87 × 10−4 | 1.54 × 10−10 |
| G73 | 1.16 × 106 | 3.41 × 10−4 | 2.94 × 10−10 |
| A15 | 7.9 × 105 | 2.5 × 10−4 | 3.17 × 10−10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhang, R.; Zhong, K.; Guo, W.; Tong, A. An Anti-CD7 Antibody–Drug Conjugate Target Showing Potent Antitumor Activity for T-Lymphoblastic Leukemia (T-ALL). Biomolecules 2024, 14, 106. https://doi.org/10.3390/biom14010106
Wang S, Zhang R, Zhong K, Guo W, Tong A. An Anti-CD7 Antibody–Drug Conjugate Target Showing Potent Antitumor Activity for T-Lymphoblastic Leukemia (T-ALL). Biomolecules. 2024; 14(1):106. https://doi.org/10.3390/biom14010106
Chicago/Turabian StyleWang, Shiqi, Ruyuan Zhang, Kunhong Zhong, Wenhao Guo, and Aiping Tong. 2024. "An Anti-CD7 Antibody–Drug Conjugate Target Showing Potent Antitumor Activity for T-Lymphoblastic Leukemia (T-ALL)" Biomolecules 14, no. 1: 106. https://doi.org/10.3390/biom14010106
APA StyleWang, S., Zhang, R., Zhong, K., Guo, W., & Tong, A. (2024). An Anti-CD7 Antibody–Drug Conjugate Target Showing Potent Antitumor Activity for T-Lymphoblastic Leukemia (T-ALL). Biomolecules, 14(1), 106. https://doi.org/10.3390/biom14010106