

Casein Kinase 1 Phosphomimetic Mutations Negatively Impact Connexin-43 Gap Junctions in Human Pluripotent Stem Cell-Derived Cardiomyocytes
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
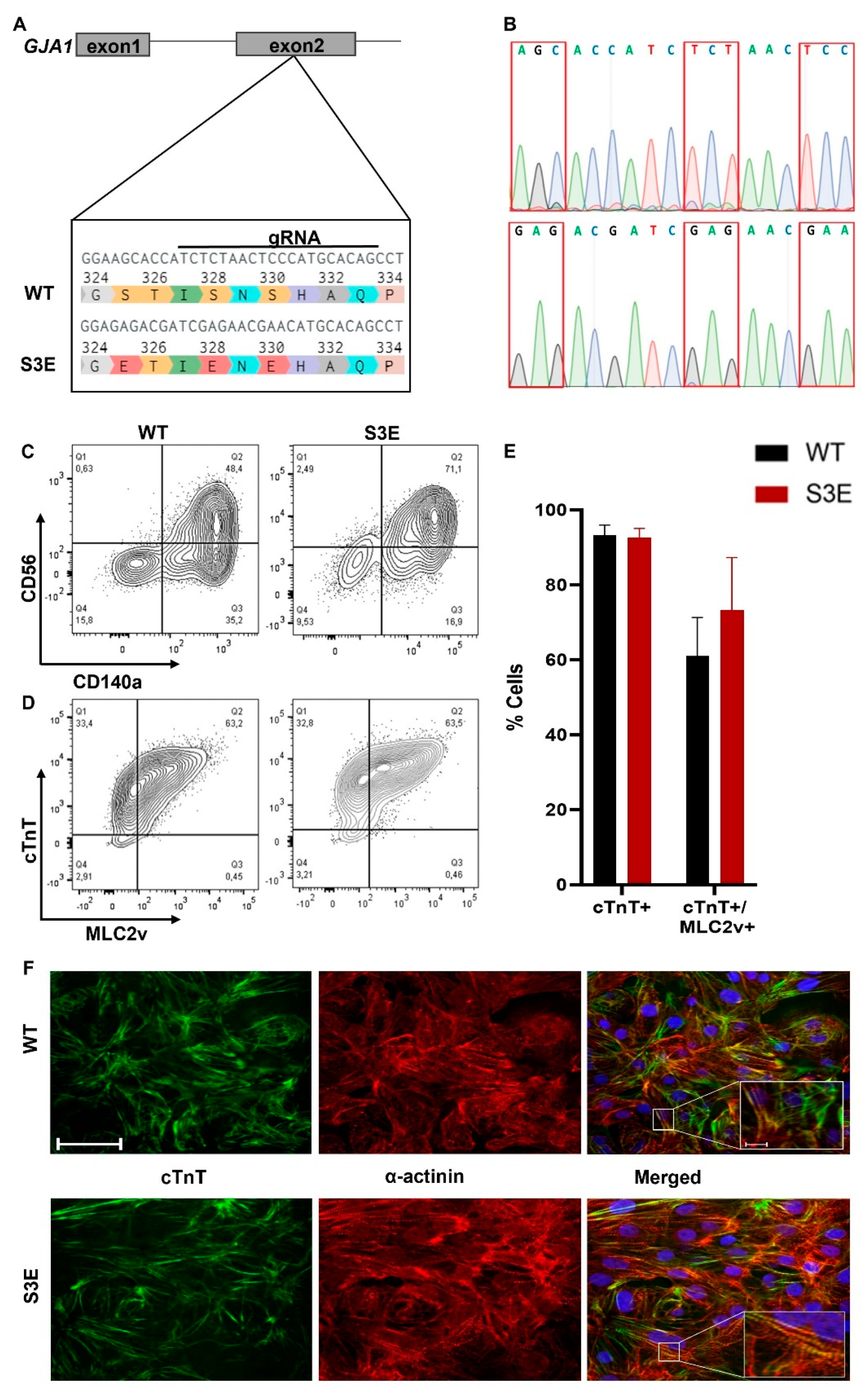
2.1. Generation of Transgenic Cx43-S3E hPSCs
2.2. Cardiac Differentiation of hPSCs
2.3. Flow Cytometry for Mesodermal and Cardiac Markers
2.4. Immunofluorescence Staining
2.5. Fluorescent Voltage and Calcium Imaging
2.6. Western Blot
2.7. Gene Expression
2.8. Statistical Analysis
3. Results
3.1. Generation and Characterization of Cx43-S3E hPSCs
3.2. Cardiac Differentiation of Cx43-S3E hPSCs
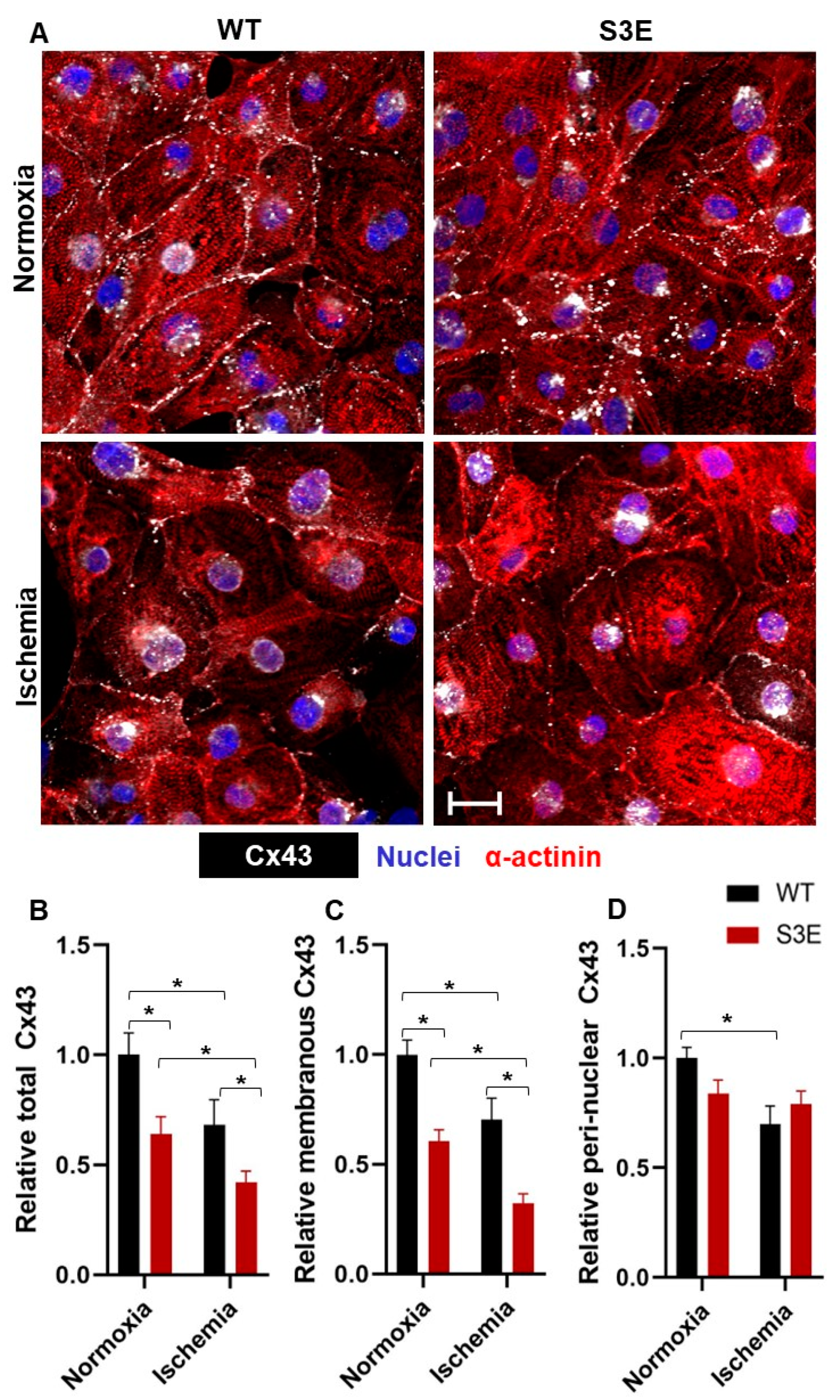
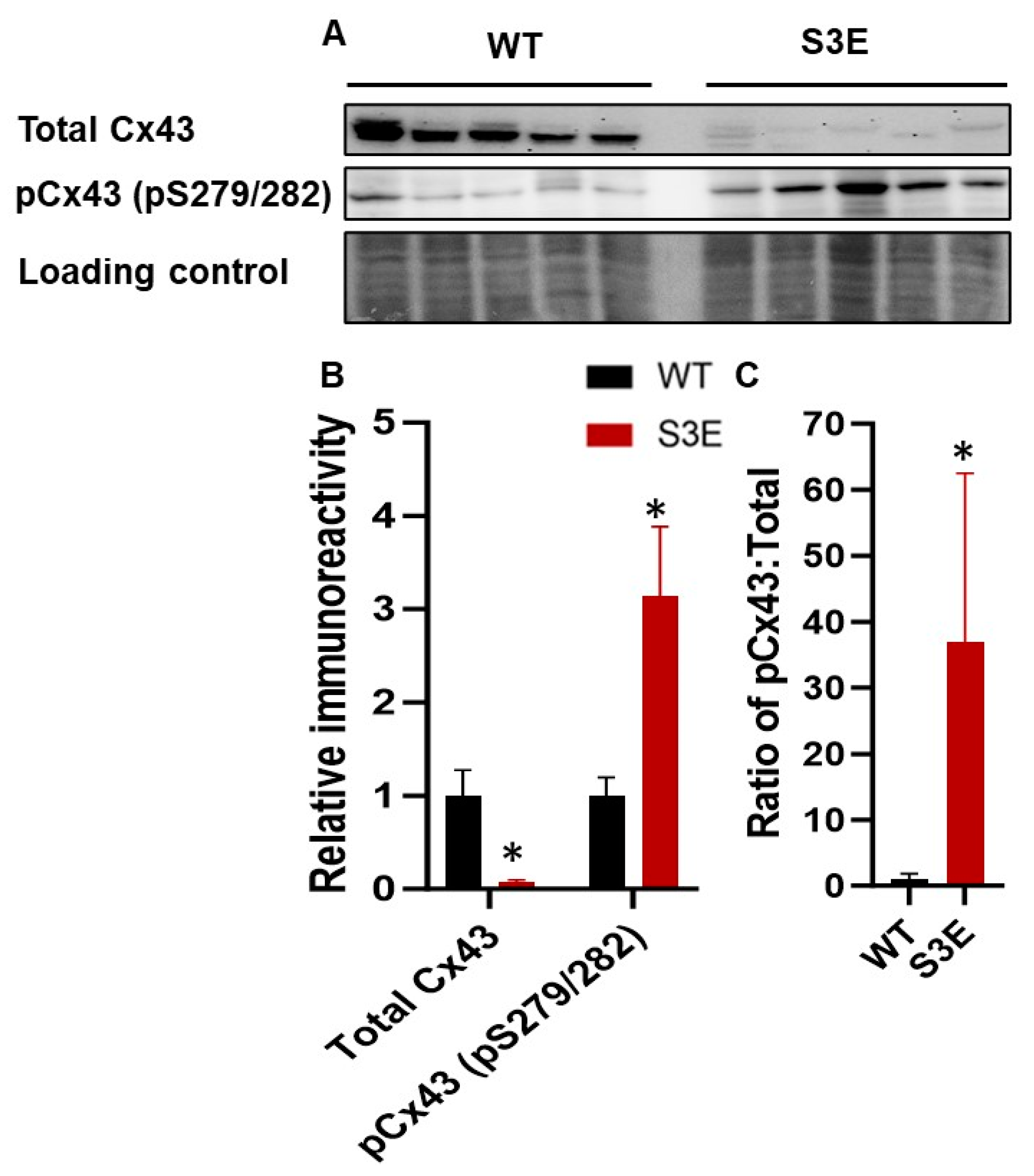
3.3. Cx43 Expression and Localization in Cx43-S3E versus WT hPSC-CMs
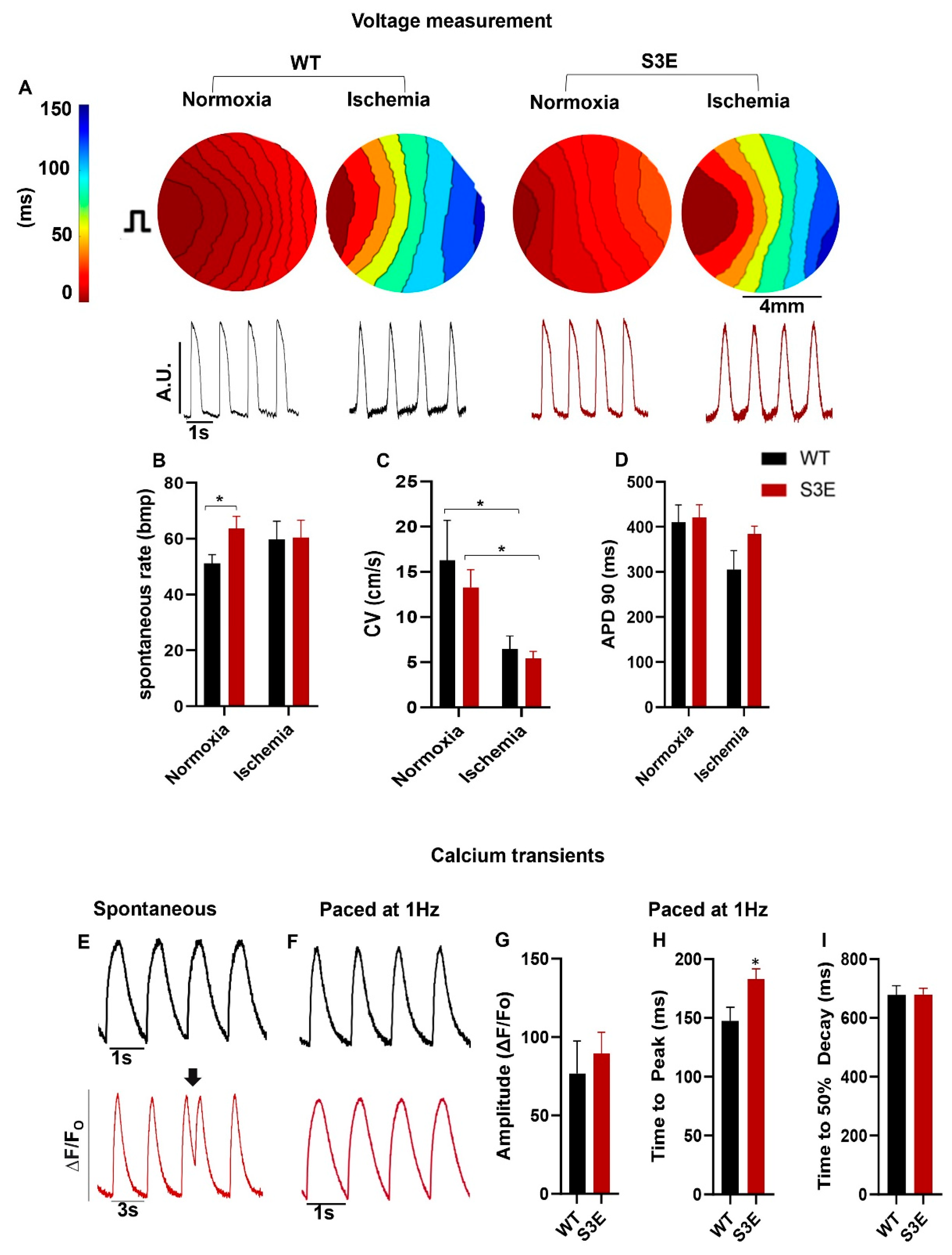
3.4. Electrophysiology and Intracellular Calcium Handling in Cx43-S3E versus WT hPSC-CMs
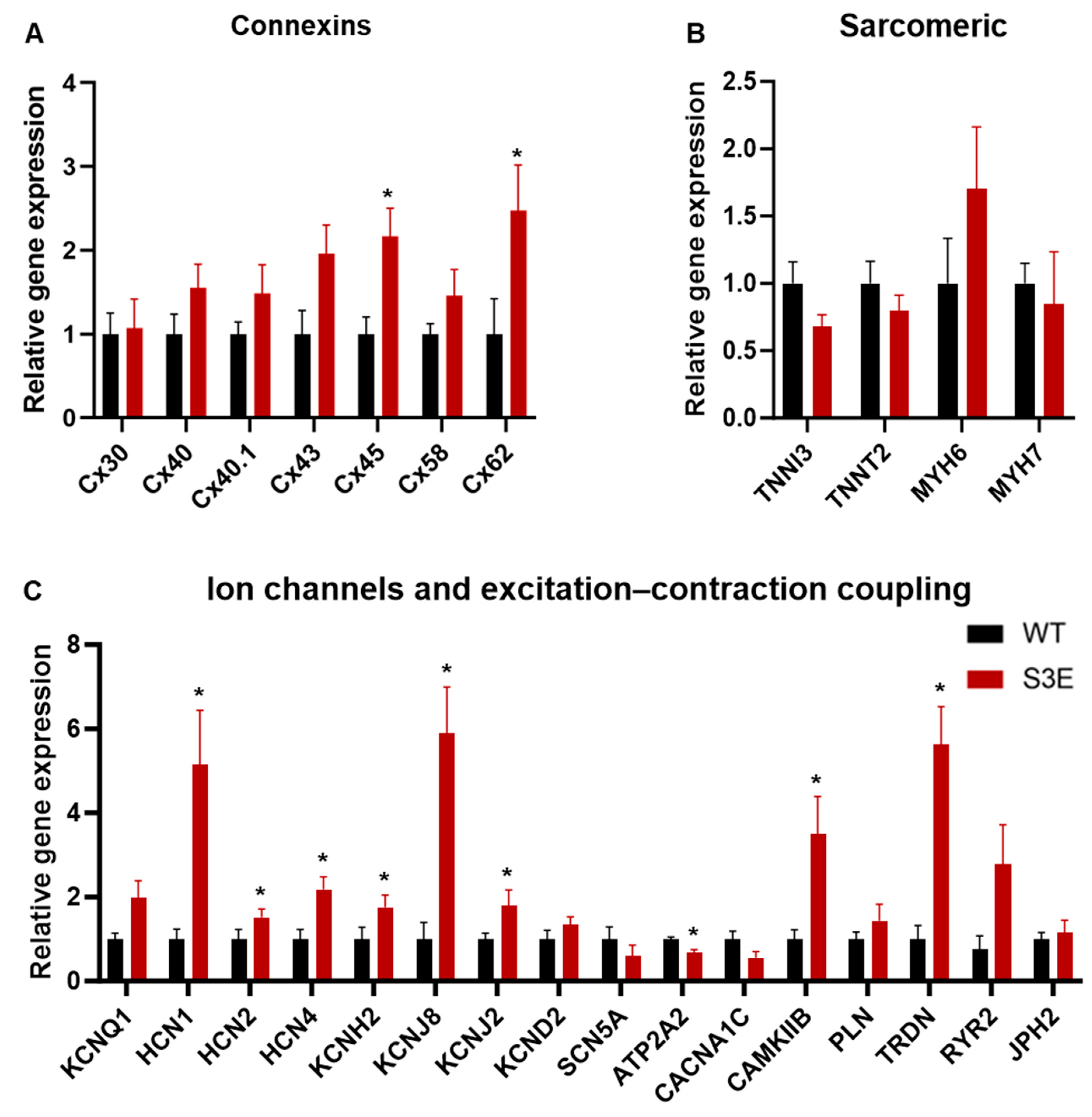
3.5. Other Transcriptional Changes in Cx43-S3E vs. WT hPSC-CMs
3.6. Status of Other Cx43 Phosphorylation Sites in Cx43-S3E vs. WT hPSC-CMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| hPSC | human pluripotent stem cell |
| hPSC-CM | human pluripotent stem cell-derived cardiomyocyte |
| Cx43 | connexin 43 |
| GJ | gap junction |
| MI | myocardial infarction |
| AP | action potential |
| APD | action potential duration |
| CK1 | casein kinase 1 |
| PKC | protein kinase C |
| MAPK | mitogen-activated protein kinase |
| qRT-PCR | quantitative real-time reverse transcription polymerase chain reaction |
References
- Laflamme, M.A.; Murry, C.E. Heart Regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Shiba, Y.; Fernandes, S.; Zhu, W.-Z.; Filice, D.; Muskheli, V.; Kim, J.; Palpant, N.J.; Gantz, J.; Moyes, K.W.; Reinecke, H.; et al. Human ES-Cell-Derived Cardiomyocytes Electrically Couple and Suppress Arrhythmias in Injured Hearts. Nature 2012, 489, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Shiba, Y.; Filice, D.; Fernandes, S.; Minami, E.; Dupras, S.K.; Van Biber, B.; Trinh, P.; Hirota, Y.; Gold, J.D.; Viswanathan, M.; et al. Electrical Integration of Human Embryonic Stem Cell-Derived Cardiomyocytes in a Guinea Pig Chronic Infarct Model. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Filice, D.; Dhahri, W.; Solan, J.L.; Lampe, P.D.; Steele, E.; Milani, N.; Van Biber, B.; Zhu, W.-Z.; Valdman, T.S.; Romagnuolo, R.; et al. Optical Mapping of Human Embryonic Stem Cell-Derived Cardiomyocyte Graft Electrical Activity in Injured Hearts. Stem Cell Res. Ther. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Dhahri, W.; Sadikov Valdman, T.; Wilkinson, D.; Pereira, E.; Ceylan, E.; Andharia, N.; Qiang, B.; Masoudpour, H.; Wulkan, F.; Quesnel, E.; et al. In Vitro Matured Human Pluripotent Stem Cell–Derived Cardiomyocytes Form Grafts with Enhanced Structure and Function in Injured Hearts. Circulation 2022, 145, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.-W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human Embryonic-Stem-Cell-Derived Cardiomyocytes Regenerate Non-Human Primate Hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Human Embryonic Stem Cell–Derived Cardiomyocytes Restore Function in Infarcted Hearts of Non-Human Primates. Nat. Biotechnol. 2018, 36, 597–605. [Google Scholar] [CrossRef]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic Transplantation of iPS Cell-Derived Cardiomyocytes Regenerates Primate Hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Gold, J.; Xu, C.; Hassanipour, M.; Rosler, E.; Police, S.; Muskheli, V.; Murry, C.E. Formation of Human Myocardium in the Rat Heart from Human Embryonic Stem Cells. Am. J. Pathol. 2005, 167, 663–671. [Google Scholar] [CrossRef]
- Funakoshi, S.; Miki, K.; Takaki, T.; Okubo, C.; Hatani, T.; Chonabayashi, K.; Nishikawa, M.; Takei, I.; Oishi, A.; Narita, M.; et al. Enhanced Engraftment, Proliferation and Therapeutic Potential in Heart Using Optimized Human iPSC-Derived Cardiomyocytes. Sci. Rep. 2016, 6, 19111. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes Derived from Human Embryonic Stem Cells in Pro-Survival Factors Enhance Function of Infarcted Rat Hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. [Google Scholar] [CrossRef]
- Agullo-Pascual, E.; Delmar, M. The Noncanonical Functions of Cx43 in the Heart. J. Membr. Biol. 2012, 245, 477–482. [Google Scholar] [CrossRef]
- Matsuyama, D.; Kawahara, K. Proliferation of Neonatal Cardiomyocytes by Connexin43 Knockdown via Synergistic Inactivation of P38 MAPK and Increased Expression of FGF1. Basic Res. Cardiol. 2009, 104, 631–642. [Google Scholar] [CrossRef]
- Smyth, J.W.; Shaw, R.M. Autoregulation of Connexin43 Gap Junction Formation by Internally Translated Isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The Connexin 43 C-Terminus: A Tail of Many Tales. Biochim. Biophys. Acta—Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap Junction Protein Connexin-43 Is a Direct Transcriptional Regulator of N-Cadherin in Vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef]
- Salat-Canela, C.; Sesé, M.; Peula, C.; Ramón y Cajal, S.; Aasen, T. Internal Translation of the Connexin 43 Transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef]
- Calderón, J.F.; Retamal, M.A. Regulation of Connexins Expression Levels by MicroRNAs, an Update. Front. Physiol. 2016, 7, 558. [Google Scholar] [CrossRef]
- Curcio, A.; Torella, D.; Iaconetti, C.; Pasceri, E.; Sabatino, J.; Sorrentino, S.; Giampà, S.; Micieli, M.; Polimeni, A.; Henning, B.J.; et al. MicroRNA-1 Downregulation Increases Connexin 43 Displacement and Induces Ventricular Tachyarrhythmias in Rodent Hypertrophic Hearts. PLoS ONE 2013, 8, e70158. [Google Scholar] [CrossRef]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of Connexin43 by MicroRNA-130a in Cardiomyocytes Results in Cardiac Arrhythmias. J. Mol. Cell. Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef]
- Bian, B.; Yu, X.-F.; Wang, G.-Q.; Teng, T.-M. Role of MiRNA-1 in Regulating Connexin 43 in Ischemia–Reperfusion Heart Injury: A Rat Model. Cardiovasc. Pathol. 2017, 27, 37–42. [Google Scholar] [CrossRef]
- Wahl, C.-M.; Schmidt, C.; Hecker, M.; Ullrich, N.D. Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases. Int. J. Mol. Sci. 2022, 23, 10174. [Google Scholar] [CrossRef]
- Kjenseth, A.; Fykerud, T.A.; Sirnes, S.; Bruun, J.; Yohannes, Z.; Kolberg, M.; Omori, Y.; Rivedal, E.; Leithe, E. The Gap Junction Channel Protein Connexin 43 is Covalently Modified and Regulated by SUMOylation. J. Biol. Chem. 2012, 287, 15851–15861. [Google Scholar] [CrossRef]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A.; et al. Nε-Lysine Acetylation Determines Dissociation from GAP Junctions and Lateralization of Connexin 43 in Normal and Dystrophic Heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of Connexin43 on Serine368 by Protein Kinase C Regulates Gap Junctional Communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Hirschhäuser, C.; Lissoni, A.; Görge, P.M.; Lampe, P.D.; Heger, J.; Schlüter, K.-D.; Leybaert, L.; Schulz, R.; Boengler, K. Connexin 43 Phosphorylation by Casein Kinase 1 is Essential for the Cardioprotection by Ischemic Preconditioning. Basic Res. Cardiol. 2021, 116, 21. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Spatio-Temporal Regulation of Connexin43 Phosphorylation and Gap Junction Dynamics. Biochim. Biophys. Acta—Biomembr. 2018, 1860, 83–90. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Specific Cx43 Phosphorylation Events Regulate Gap Junction Turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef]
- Imanaga, I.; Hai, L.; Ogawa, K.; Matsumura, K.; Mayama, T. Phosphorylation of Connexin in Functional Regulation of the Cardiac Gap Junction. Exp. Clin. Cardiol. 2004, 9, 161–164. [Google Scholar]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L.; et al. Compartmentalized Connexin 43 S -Nitrosylation/Denitrosylation Regulates Heterocellular Communication in the Vessel Wall. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-Nitrosylation and Permeation through Connexin 43 Hemichannels in Astrocytes: Induction by Oxidant Stress and Reversal by Reducing Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef]
- Leithe, E.; Kjenseth, A.; Sirnes, S.; Stenmark, H.; Brech, A.; Rivedal, E. Ubiquitylation of the Gap Junction Protein Connexin-43 Signals Its Trafficking from Early Endosomes to Lysosomes in a Process Mediated by Hrs and Tsg101. J. Cell Sci. 2009, 122, 3883–3893. [Google Scholar] [CrossRef]
- Falk, M.M.; Kells, R.M.; Berthoud, V.M. Degradation of Connexins and Gap Junctions. FEBS Lett. 2014, 588, 1221–1229. [Google Scholar] [CrossRef]
- Solan, J.L.; Márquez-Rosado, L.; Lampe, P.D. Cx43 Phosphorylation–Mediated Effects on ERK and Akt Protect against Ischemia Reperfusion Injury and Alter the Stability of the Stress-Inducible Protein NDRG1. J. Biol. Chem. 2019, 294, 11762–11771. [Google Scholar] [CrossRef]
- Lampe, P.D.; Lau, A.F. The Effects of Connexin Phosphorylation on Gap Junctional Communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Li, H.; Spagnol, G.; Zheng, L.; Stauch, K.L.; Sorgen, P.L. Regulation of Connexin43 Function and Expression by Tyrosine Kinase 2. J. Biol. Chem. 2016, 291, 15867–15880. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 Phosphorylation: Structural Changes and Biological Effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef]
- Turner, M.S.; Haywood, G.A.; Andreka, P.; You, L.; Martin, P.E.; Evans, W.H.; Webster, K.A.; Bishopric, N.H. Reversible Connexin 43 Dephosphorylation During Hypoxia and Reoxygenation is Linked to Cellular ATP Levels. Circ. Res. 2004, 95, 726–733. [Google Scholar] [CrossRef]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of Connexin43 Phosphorylated at S325, S328 and S330 in Normoxic and Ischemic Heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef]
- Remo, B.F.; Qu, J.; Volpicelli, F.M.; Giovannone, S.; Shin, D.; Lader, J.; Liu, F.; Zhang, J.; Lent, D.S.; Morley, G.E.; et al. Phosphatase-Resistant Gap Junctions Inhibit Pathological Remodeling and Prevent Arrhythmias. Circ. Res. 2011, 108, 1459–1466. [Google Scholar] [CrossRef]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Gonzalez, J.P.; Zhao, Q.; Xie, L.-H.; Li, H.; Liu, T.; Wehrens, X.H.T.; Lampe, P.D.; et al. Prevention of Connexin-43 Remodeling Protects against Duchenne Muscular Dystrophy Cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1727. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Crook, J.M.; Peura, T.T.; Kravets, L.; Bosman, A.G.; Buzzard, J.J.; Horne, R.; Hentze, H.; Dunn, N.R.; Zweigerdt, R.; Chua, F.; et al. The Generation of Six Clinical-Grade Human Embryonic Stem Cell Lines. Cell Stem Cell 2007, 1, 490–494. [Google Scholar] [CrossRef]
- Funk, W.D.; Labat, I.; Sampathkumar, J.; Gourraud, P.-A.; Oksenberg, J.R.; Rosler, E.; Steiger, D.; Sheibani, N.; Caillier, S.; Stache-Crain, B.; et al. Evaluating the Genomic and Sequence Integrity of Human ES Cell Lines; Comparison to Normal Genomes. Stem Cell Res. 2012, 8, 154–164. [Google Scholar] [CrossRef]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human Cardiovascular Progenitor Cells Develop from a KDR+ Embryonic-Stem-Cell-Derived Population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Yang, D.; Gomez-Garcia, J.; Funakoshi, S.; Tran, T.; Fernandes, I.; Bader, G.D.; Laflamme, M.A.; Keller, G.M. Modeling Human Multi-Lineage Heart Field Development with Pluripotent Stem Cells. Cell Stem Cell 2022, 29, 1382–1401.e8. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid Turnover of Connexin43 in the Adult Rat Heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef]
- Saffitz, J.E.; Laing, J.G.; Yamada, K.A. Connexin Expression and Turnover. Circ. Res. 2000, 86, 723–728. [Google Scholar] [CrossRef]
- O’Shea, C.; Holmes, A.P.; Yu, T.Y.; Winter, J.; Wells, S.P.; Correia, J.; Boukens, B.J.; De Groot, J.R.; Chu, G.S.; Li, X.; et al. ElectroMap: High-Throughput Open-Source Software for Analysis and Mapping of Cardiac Electrophysiology. Sci. Rep. 2019, 9, 1389. [Google Scholar] [CrossRef]
- Al-attar, R.; Storey, K.B. RAGE Management: ETS1- EGR1 Mediated Transcriptional Networks Regulate Angiogenic Factors in Wood Frogs. Cell. Signal. 2022, 98, 110408. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 in LA-25 Cells with Active v-Src is Phosphorylated on Y247, Y265, S262, S279/282, and S368 via Multiple Signaling Pathways. Cell Commun. Adhes. 2008, 15, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Meng, Q.; Yu, X.; Jing, X.; Xu, P.; Luo, D. Regulatory Effect of Connexin 43 on Basal Ca2+ Signaling in Rat Ventricular Myocytes. PLoS ONE 2012, 7, e36165. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Doble, B.W.; Kardami, E. The Carboxy-Tail of Connexin-43 Localizes to the Nucleus and Inhibits Cell Growth. Mol. Cell. Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of Connexin-43 Gap Junctional Intercellular Communication by Mitogen-Activated Protein Kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.; Singh, R.; Wang, Z.Z. Arrhythmogenic Risks of Stem Cell Replacement Therapy for Cardiovascular Diseases. J. Cell. Physiol. 2020, 235, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.E.; Marchianó, S.; Zhang, K.; Yang, X.; Murry, C.E.; Boyle, P.M. Graft–Host Coupling Changes Can Lead to Engraftment Arrhythmia: A Computational Study. J. Physiol. 2023, 601, 2733–2749. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Siegelbaum, S.A. Properties of Hyperpolarization-Activated Pacemaker Current Defined by Coassembly of Hcn1 and Hcn2 Subunits and Basal Modulation by Cyclic Nucleotide. J. Gen. Physiol. 2001, 117, 491–504. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Burashnikov, A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card. Electrophysiol. Clin. 2011, 3, 23–45. [Google Scholar] [CrossRef]
- Tomek, J.; Tomková, M.; Zhou, X.; Bub, G.; Rodriguez, B. Modulation of Cardiac Alternans by Altered Sarcoplasmic Reticulum Calcium Release: A Simulation Study. Front. Physiol. 2018, 9, 1306. [Google Scholar] [CrossRef]
- Gomez-Garcia, M.J.; Quesnel, E.; Al-attar, R.; Laskary, A.R.; Laflamme, M.A. Maturation of Human Pluripotent Stem Cell Derived Cardiomyocytes in vitro and in vivo. Semin. Cell Dev. Biol. 2021, 118, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Garay, B.I.; Givens, S.; Abreu, P.; Liu, M.; Yücel, D.; Baik, J.; Stanis, N.; Rothermel, T.M.; Magli, A.; Abrahante, J.E.; et al. Dual Inhibition of MAPK and PI3K/AKT Pathways Enhances Maturation of Human iPSC-Derived Cardiomyocytes. Stem Cell Rep. 2022, 17, 2005–2022. [Google Scholar] [CrossRef] [PubMed]
- King, T.J.; Lampe, P.D. Temporal Regulation of Connexin Phosphorylation in Embryonic and Adult Tissues. Biochim. Biophys. Acta—Biomembr. 2005, 1719, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, J.A.; Valdez, S.; Taylor, L.; Whisenant, C.; Selzman, C.H.; Drakos, S.G.; Ranjan, R.; Hong, T.; Saffitz, J.E.; Shaw, R.M. GJA1-20k Rescues Cx43 Localization and Arrhythmias in Arrhythmogenic Cardiomyopathy. Circ. Res. 2023, 132, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Hong, T.-T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.R.; Chi, N.C.; Shaw, R.M. Limited Forward Trafficking of Connexin 43 Reduces Cell-Cell Coupling in Stressed Human and Mouse Myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary Trafficking Subunit GJA1-20k Protects Connexin-43 from Degradation and Limits Ventricular Arrhythmias. J. Clin. Investig. 2020, 130, 4858–4870. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.D.; Zhu, W.-Z.; Regnier, M.; Laflamme, M.A. Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef]
- Carson, D.; Hnilova, M.; Yang, X.; Nemeth, C.L.; Tsui, J.H.; Smith, A.S.T.; Jiao, A.; Regnier, M.; Murry, C.E.; Tamerler, C.; et al. Nanotopography-Induced Structural Anisotropy and Sarcomere Development in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells. ACS Appl. Mater. Interfaces 2016, 8, 21923–21932. [Google Scholar] [CrossRef]
- Funakoshi, S.; Fernandes, I.; Mastikhina, O.; Wilkinson, D.; Tran, T.; Dhahri, W.; Mazine, A.; Yang, D.; Burnett, B.; Lee, J.; et al. Generation of Mature Compact Ventricular Cardiomyocytes from Human Pluripotent Stem Cells. Nat. Commun. 2021, 12, 3155. [Google Scholar] [CrossRef]
- Yoshida, S.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ohashi, F.; Toyofuku, T.; Toda, K.; Sawa, Y. Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes by Soluble Factors from Human Mesenchymal Stem Cells. Mol. Ther. 2018, 26, 2681–2695. [Google Scholar] [CrossRef]
- Miklas, J.W.; Nunes, S.S.; Sofla, A.; Reis, L.A.; Pahnke, A.; Xiao, Y.; Laschinger, C.; Radisic, M. Bioreactor for Modulation of Cardiac Microtissue Phenotype by Combined Static Stretch and Electrical Stimulation. Biofabrication 2014, 6, 024113. [Google Scholar] [CrossRef] [PubMed]
- Kensah, G.; Roa Lara, A.; Dahlmann, J.; Zweigerdt, R.; Schwanke, K.; Hegermann, J.; Skvorc, D.; Gawol, A.; Azizian, A.; Wagner, S.; et al. Murine and Human Pluripotent Stem Cell-Derived Cardiac Bodies Form Contractile Myocardial Tissue in vitro. Eur. Heart J. 2013, 34, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Wustmann, A.; Karl, S.; Blanke, K.; Apel, D.; Rojas-Gomez, D.; Franke, H.; Mohr, F.W.; Janousek, J.; Dhein, S. Cyclic Mechanical Stretch Induces Cardiomyocyte Orientation and Polarization of the Gap Junction Protein Connexin43. Circ. Res. 2010, 106, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.-L.; Tulloch, N.L.; Saiget, M.; Paige, S.L.; Razumova, M.V.; Regnier, M.; Tung, K.C.; Keller, G.; Pabon, L.; Reinecke, H.; et al. Mechanical Stress Promotes Maturation of Human Myocardium From Pluripotent Stem Cell-Derived Progenitors. Stem Cells 2015, 33, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- LaBarge, W.; Mattappally, S.; Kannappan, R.; Fast, V.G.; Pretorius, D.; Berry, J.L.; Zhang, J. Maturation of Three-Dimensional, HiPSC-Derived Cardiomyocyte Spheroids Utilizing Cyclic, Uniaxial Stretch and Electrical Stimulation. PLoS ONE 2019, 14, e0219442. [Google Scholar] [CrossRef]
- Morley, G.E.; Vaidya, D.; Samie, F.H.; Lo, C.; Delmar, M.; Jalife, J. Characterization of Conduction in the Ventricles of Normal and Heterozygous Cx43 Knockout Mice using Optical Mapping. J. Cardiovasc. Electrophysiol. 1999, 10, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of Cardiac Gap Junction Expression and Arrhythmic Susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef]
- Sánchez, J.A.; Rodríguez-Sinovas, A.; Fernández-Sanz, C.; Ruiz-Meana, M.; García-Dorado, D. Effects of a Reduction in the Number of Gap Junction Channels or in Their Conductance on Ischemia-Reperfusion Arrhythmias in Isolated Mouse Hearts. Am. J. Physiol. Circ. Physiol. 2011, 301, H2442–H2453. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythmia Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef]
- van Veen, T.A.B.; Stein, M.; Royer, A.; Le Quang, K.; Charpentier, F.; Colledge, W.H.; Huang, C.L.-H.; Wilders, R.; Grace, A.A.; Escande, D.; et al. Impaired Impulse Propagation in Scn5a-Knockout Mice. Circulation 2005, 112, 1927–1935. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Electrophysiological Effects of Remodeling Cardiac Gap Junctions and Cell Size. Circ. Res. 2000, 86, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.B.; Veeraraghavan, R.; Poelzing, S.; Weinberg, S.H. Cellular Size, Gap Junctions, and Sodium Channel Properties Govern Developmental Changes in Cardiac Conduction. Front. Physiol. 2021, 12, 731025. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, F.; Benzoni, P.; Campostrini, G.; Milanesi, R.; Bucchi, A.; Baruscotti, M.; Dell’Era, P.; Rossini, A.; Barbuti, A. A Detailed Characterization of the Hyperpolarization-Activated “Funny” Current (If) in Human-Induced Pluripotent Stem Cell (iPSC)–Derived Cardiomyocytes with Pacemaker Activity. Pflügers Arch.—Eur. J. Physiol. 2021, 473, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Yang, L.; Lin, B.; Zhu, X.; Sun, B.; Kaplan, A.D.; Bett, G.C.L.; Rasmusson, R.L.; London, B.; Salama, G. Mechanism of Automaticity in Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. J. Mol. Cell. Cardiol. 2015, 81, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human IPSC-Derived Cardiomyocytes: In silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Biktashev, V.N.; Arutunyan, A.; Sarvazyan, N.A. Generation and Escape of Local Waves from the Boundary of Uncoupled Cardiac Tissue. Biophys. J. 2008, 94, 3726–3738. [Google Scholar] [CrossRef] [PubMed]
- Sottas, V.; Wahl, C.-M.; Trache, M.C.; Bartolf-Kopp, M.; Cambridge, S.; Hecker, M.; Ullrich, N.D. Improving Electrical Properties of iPSC-Cardiomyocytes by Enhancing Cx43 Expression. J. Mol. Cell. Cardiol. 2018, 120, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Smit, N.W.; Coronel, R. Stem Cells Can form Gap Junctions with Cardiac Myocytes and Exert Pro-Arrhythmic Effects. Front. Physiol. 2014, 5, 419. [Google Scholar] [CrossRef]
- Johnson, K.E.; Mitra, S.; Katoch, P.; Kelsey, L.S.; Johnson, K.R.; Mehta, P.P. Phosphorylation on Ser-279 and Ser-282 of Connexin43 Regulates Endocytosis and Gap Junction Assembly in Pancreatic Cancer Cells. Mol. Biol. Cell 2013, 24, 715–733. [Google Scholar] [CrossRef]
- Matsuda, T.; Fujio, Y.; Nariai, T.; Ito, T.; Yamane, M.; Takatani, T.; Takahashi, K.; Azuma, J. N-Cadherin Signals through Rac1 Determine the Localization of Connexin 43 in Cardiac Myocytes. J. Mol. Cell. Cardiol. 2006, 40, 495–502. [Google Scholar] [CrossRef]
- Lin, R.; Warn-Cramer, B.J.; Kurata, W.E.; Lau, A.F. V-Src Phosphorylation of Connexin 43 on Tyr247 and Tyr265 Disrupts Gap Junctional Communication. J. Cell Biol. 2001, 154, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, W.; Luo, G.; Alain, L.A. Hypoxia Induces Connexin 43 Dysregulation by Modulating Matrix Metalloproteinases via MAPK Signaling. Mol. Cell. Biochem. 2013, 384, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Xiao, Y.; Song, X.; Kang, Y.J. Extracellular Matrix Remodeling is Associated with the Survival of Cardiomyocytes in the Subendocardial Region of the Ischemic Myocardium. Exp. Biol. Med. 2021, 246, 2579–2588. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-attar, R.; Jargstorf, J.; Romagnuolo, R.; Jouni, M.; Alibhai, F.J.; Lampe, P.D.; Solan, J.L.; Laflamme, M.A. Casein Kinase 1 Phosphomimetic Mutations Negatively Impact Connexin-43 Gap Junctions in Human Pluripotent Stem Cell-Derived Cardiomyocytes. Biomolecules 2024, 14, 61. https://doi.org/10.3390/biom14010061
Al-attar R, Jargstorf J, Romagnuolo R, Jouni M, Alibhai FJ, Lampe PD, Solan JL, Laflamme MA. Casein Kinase 1 Phosphomimetic Mutations Negatively Impact Connexin-43 Gap Junctions in Human Pluripotent Stem Cell-Derived Cardiomyocytes. Biomolecules. 2024; 14(1):61. https://doi.org/10.3390/biom14010061
Chicago/Turabian StyleAl-attar, Rasha, Joseph Jargstorf, Rocco Romagnuolo, Mariam Jouni, Faisal J. Alibhai, Paul D. Lampe, Joell L. Solan, and Michael A. Laflamme. 2024. "Casein Kinase 1 Phosphomimetic Mutations Negatively Impact Connexin-43 Gap Junctions in Human Pluripotent Stem Cell-Derived Cardiomyocytes" Biomolecules 14, no. 1: 61. https://doi.org/10.3390/biom14010061
APA StyleAl-attar, R., Jargstorf, J., Romagnuolo, R., Jouni, M., Alibhai, F. J., Lampe, P. D., Solan, J. L., & Laflamme, M. A. (2024). Casein Kinase 1 Phosphomimetic Mutations Negatively Impact Connexin-43 Gap Junctions in Human Pluripotent Stem Cell-Derived Cardiomyocytes. Biomolecules, 14(1), 61. https://doi.org/10.3390/biom14010061