
Factors and Mechanisms of Thyroid Hormone Activity in the Brain: Possible Role in Recovery and Protection



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. THs and Normal Brain Development
3. Nuclear TH Receptors
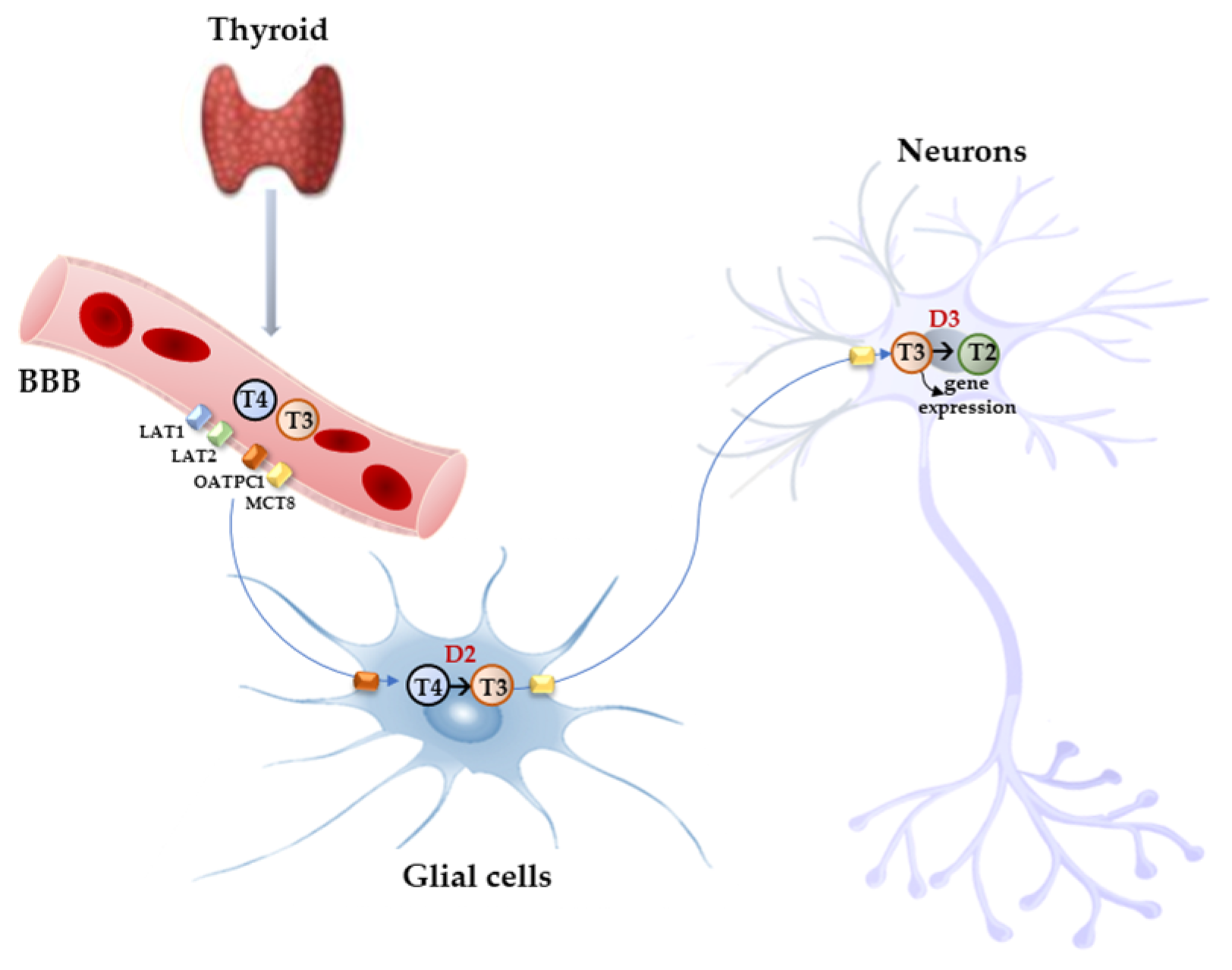
4. TH Transporters
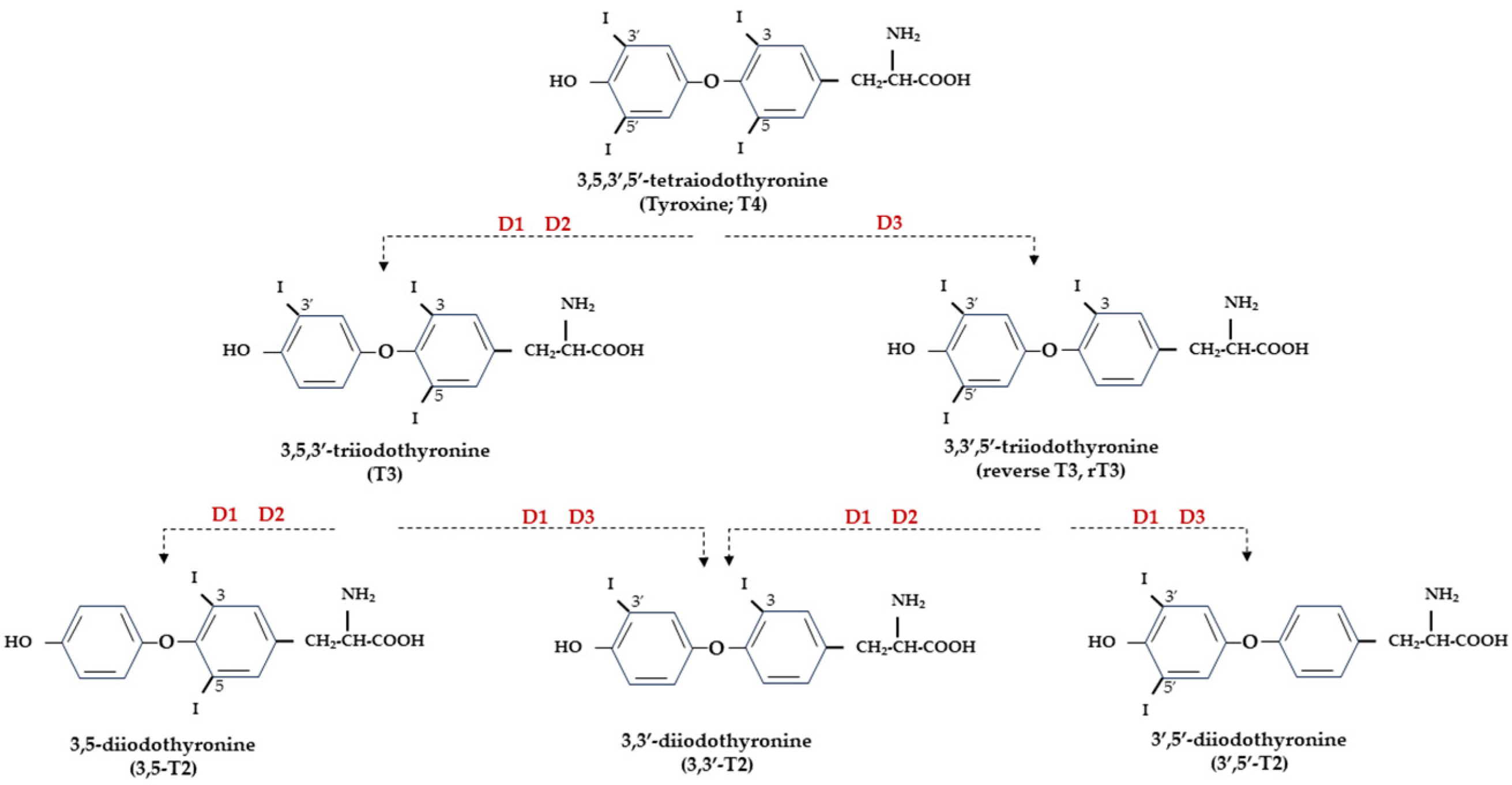
5. TH Deiodinases
6. THs and Experimental Models of Brain Injury
7. THs and In Vivo Models of Cognitive/Emotional Impairment
8. THs and Alzheimer’s Disease
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Porterfield, S.P.; Hendrich, C.E. The role of thyroid hormones in prenatal and neonatal neurological development—Current perspectives. Endocr. Rev. 1993, 14, 94–106. [Google Scholar]
- Oppenheimer, J.H.; Schwartz, H.L. Molecular basis of thyroid hormone-dependent brain development. Endocr. Rev. 1997, 18, 462–475. [Google Scholar] [PubMed]
- de Escobar, G.M.; Obregon, M.J.; del Rey, F.E. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract. Res. Clin. Endocrinol. Metabol. 2004, 18, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Thoemke, K.R.; Anderson, G.W. The role of thyroid hormone in fetal and neonatal brain development. Curr. Opin. Endocrinol. Diabetes 2005, 12, 10–16. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef] [PubMed]
- Wirth, E.K.; Schweizer, U.; Köhrle, J. Transport of thyroid hormone in brain. Front. Endocrinol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Thyroid hormone transporters in health and disease: Advances in thyroid hormone deiodination. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, L.; Vassalle, C.; Iervasi, G.; Sabatino, L. Main Factors Involved in Thyroid Hormone Action. Molecules 2021, 26, 7337. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Vassalle, C.; Del Seppia, C.; Iervasi, G. Deiodinases and the Three Types of Thyroid Hormone Deiodination Reactions. Endocrinol. Metab. 2021, 36, 952–964. [Google Scholar] [CrossRef]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeold, A.; Bianco, A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Leonard, J.L.; Davis, F.B. Mechanisms of nongenomic actions of thyroid hormone. Front. Neuroendocrinol. 2008, 29, 211–218. [Google Scholar] [CrossRef]
- Mousa, S.A.; O’Connor, L.; Davis, F.B.; Davis, P.J. Proangiogenesis action of the thyroid hormone analog 3,5-diiodothyropropionic acid (ditpa) is initiated at the cell surface and is integrin mediated. Endocrinology 2006, 147, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Schoonover, C.M.; Jones, S.A. Control of thyroid hormone action in the developing rat brain. Thyroid 2003, 13, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J. Thyroid hormones and brain development. Vitam. Horm. 2005, 71, 95–122. [Google Scholar] [CrossRef]
- Alcaide, M.A.; Mayerl, S. Local thyroid hormone action in brain development. Int. J. Mol. Sci. 2023, 24, 12352. [Google Scholar] [CrossRef]
- Miranda, A.; Sousa, N. Maternal hormonal milieu influence on fetal brain development. Brain Behav. 2018, 8, e00920. [Google Scholar] [CrossRef]
- Andersen, S.L.; Laurberg, P.; Wu, C.S.; Olsen, J. Attention deficit hyperactivity disorder and autism spectrum disorder in children born to mothers with thyroid dysfunction: A Danish nationwide cohort study. BJOG 2014, 121, 1365–1374. [Google Scholar] [CrossRef]
- Andersen, S.L.; Olsen, J.; Wu, C.S.; Laurberg, P. Psychiatric disease in late adolescence and young adulthood. Foetal programming by maternal hypothyroidism? Clin. Endocrinol. 2014, 81, 126–133. [Google Scholar] [CrossRef]
- Powell, E.M. Interneuron development and epilepsy: Early genetic defects cause long-term consequences in seizures and susceptibility. Epilepsy Curr. 2013, 13, 172–176. [Google Scholar] [CrossRef]
- Flamant, F.; Gauthier, K. Thyroid hormone receptors: The challenge of elucidating isotype-specific functions and cell-specific response. Biochim. Biophys. Acta 2013, 1830, 3900–3907. [Google Scholar] [CrossRef]
- Wallis, K.; Dudazy, S.; van Hogerlinden, M.; Nordstrom, K.; Mittag, J.; Vennstrom, B. The thyroid hormone receptor {alpha}1 protein is expressed in embryonic postmitotic neurons and persists in most adult neurons. Mol. Endocrinol. 2010, 24, 1904–1916. [Google Scholar] [CrossRef]
- Lazar, M.A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 1993, 14, 184–193. [Google Scholar]
- Liu, Y.Y.; Brent, G.A. The Role of Thyroid Hormone in Neuronal Protection. Compr. Physiol. 2021, 11, 2075–2095. [Google Scholar] [CrossRef]
- Bernal, J.; Morte, B. Thyroid hormone receptor activity in the absence of ligand: Physiological and developmental implications. Biochim. Biophys. Acta 2013, 1830, 3893–3899. [Google Scholar] [CrossRef]
- Muñoz, A.; Wrighton, C.; Seliger, B.; Bernal, J.; Beug, H. Thyroid hormone receptor/c-erbA: Control of commitment and differentiation in the neuronal/chromaffin progenitor line PC12. J. Cell Biol. 1993, 121, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.; Guadaño-Ferraz, A.; Morte, B. Thyroid hormone transport—Functions and clinical implications. Nat. Rev. Endocrinol. 2015, 11, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Schweizer, U. Thyroid Hormone Transport and Transporters. In Vitamins and Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 106, pp. 19–44. [Google Scholar] [CrossRef]
- Kinne, A.; Schülein, R.; Krause, G. Primary and secondary thyroid hormone transporters. Thyroid Res. 2011, 4, S7. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Kuiper, G.G.; Jansen, J.; Visser, T.J.; Kester, M.H. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol. Endocrinol. 2006, 20, 2761–2772. [Google Scholar] [CrossRef]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Barrett, T.; Mancilla, E.E.; Svensson, J.; Kester, M.H.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Dumitrescu, A.M.; Liao, X.H.; Best, T.B.; Brockmann, K.; Refetoff, S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet. 2004, 74, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.E.; Stevenson, R.E. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Woodford, K.; Zhou, M.; Black, D.S.; Haggerty, J.E.; Tate, E.H.; Grindstaff, K.K.; Mengesha, W.; Raman, C.; Zerangue, N. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008, 149, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Wirth, E.K.; Roth, S.; Blechschmidt, C.; Holter, S.M.; Becker, L.; Racz, I.; Zimmer, A.; Klopstock, T.; Gailus-Durner, V.; Fuchs, H.; et al. Neuronal 3’,3,5- triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J. Neurosci. 2009, 29, 9439–9449. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Jansen, J.; Jachtenberg, J.W.; Visser, W.E.; Kester, M.H.; Visser, T.J. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol. Endocrinol. 2008, 22, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, bnz008. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef]
- Heuer, H.; Maier, M.K.; Iden, S.; Mittag, J.; Friesema, E.C.; Visser, T.J.; Bauer, K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 2005, 146, 1701–1706. [Google Scholar] [CrossRef]
- Visser, W.E.; Friesema, E.C.; Jansen, J.; Visser, T.J. Thyroid hormone transport in and out of cells. Trends Endocrinol. Metab. 2008, 19, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Killian, D.M.; Chikhale, P.J. Predominant functional activity of the large, neutral amino acid transporter LAT1 isoform at the cerebrovasculature. Neurosci. Lett. 2001, 306, 1–4. [Google Scholar] [CrossRef]
- Di Cosmo, C.; Liao, X.H.; Dumitrescu, A.M.; Weiss, R.E.; Refetoff, S. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology 2009, 150, 4450–4458. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, A.M.; Liao, X.H.; Ye, H.; Weiss, R.E.; Dumitrescu, A.M.; Refetoff, S. The Thyroid Hormone Analog DITPA Ameliorates Metabolic Parameters of Male Mice with Mct8 Deficiency. Endocrinology 2015, 156, 3889–3894. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; Peeters, R.P.; Visser, T.J.; Visser, W.E. Therapeutic applications of thyroid hormone analogues in resistance to thyroid hormone (RTH) syndromes. Mol. Cell. Endocrinol. 2017, 458, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kersseboom, S.; Horn, S.; Visser, W.E.; Chen, J.; Friesema, E.C.; Vaurs-Barrière, C.; Peeters, R.P.; Heuer, H.; Visser, T.J. In Vitro and Mouse Studies Supporting Therapeutic Utility of Triiodothyroacetic Acid in MCT8 Deficiency. Mol. Endocrinol. 2014, 28, 1961–1970. [Google Scholar] [CrossRef]
- van Geest, F.S.; Gunhanlar, N.; Groeneweg, S.; Visser, W.E. Monocarboxylate Transporter 8 Deficiency: From Pathophysiological Understanding to Therapy Development. Front. Endocrinol. (Lausanne) 2021, 12, 723750. [Google Scholar] [CrossRef]
- Mayerl, S.; Müller, J.; Bauer, R.; Richert, S.; Kassmann, C.M.; Darras, V.M.; Buder, K.; Boelen, A.; Visser, T.J.; Heuer, H. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Investig. 2014, 124, 1987–1999. [Google Scholar] [CrossRef]
- Morte, B.; Gil-Ibañez, P.; Heuer, H.; Bernal, J. Brain Gene Expression in Systemic Hypothyroidism and Mouse Models of MCT8 Deficiency: The Mct8-Oatp1c1-Dio2 Triad. Thyroid 2021, 31, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; Peeters, R.P.; Moran, C.; Stoupa, A.; Auriol, F.; Tonduti, D.; Dica, A.; Paone, L.; Rozenkova, K.; Malikova, J.; et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 695–706. [Google Scholar] [CrossRef] [PubMed]
- van Geest, F.S.; Groeneweg, S.; van den Akker, E.L.T.; Bacos, I.; Barca, D.; van den Berg, S.A.A.; Bertini, E.; Brunner, D.; Brunetti-Pierri, N.; Cappa, M.; et al. Long-Term Efficacy of T3 Analogue Triac in Children and Adults with MCT8 Deficiency: A Real-Life Retrospective Cohort Study. J. Clin. Endocrinol. Metab. 2022, 107, e1136–e1147. [Google Scholar] [CrossRef]
- Köhrle, J.; Frädrich, C. Deiodinases control local cellular and systemic thyroid hormone availability. Free Radic. Biol. Med. 2022, 193, 59–79. [Google Scholar] [CrossRef]
- Gereben, B.; Zeold, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell. Mol. Life Sci. 2008, 65, 570–590. [Google Scholar] [CrossRef]
- Sabatino, L.; Iervasi, G.; Ferrazzi, P.; Francesconi, D.; Chopra, I.J. A study of iodothyronine 5′-monodeiodinase activities in normal and pathological tissues in man and their comparison with activities in rat tissues. Life Sci. 2000, 68, 191–202. [Google Scholar] [CrossRef]
- Sabatino, L.; Chopra, I.J.; Tanavoli, S.; Iacconi, P.; Iervasi, G. A radioimmunoassay for type I iodothyronine 5′-monodeiodinase in human tissues. Thyroid 2001, 11, 733–739. [Google Scholar] [CrossRef]
- Maia, A.L.; Kim, B.W.; Huang, S.A.; Harney, J.W.; Larsen, P.R. Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J. Clin. Investig. 2005, 115, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Galton, V.A.; Schneider, M.J.; Clark, A.S.; St Germain, D.L. Life without thyroxine to 3,5,3ʹ- triiodothyronine conversion: Studies in mice devoid of the 5ʹ-deiodinases. Endocrinology 2009, 150, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Pilo, A.; Iervasi, G.; Vitek, F.; Ferdeghini, M.; Cazzuola, F.; Bianchi, R. Thyroidal and peripheral production of 3,5,3′-triiodothyronine in humans by multicompartmental analysis. Am. J. Physiol. 1990, 258, E715–E726. [Google Scholar] [CrossRef] [PubMed]
- Laurberg, P.; Tørring, J.; Weeke, J. A comparison of the effects of propylthiouracil and methimazol on circulating thyroid hormones and various measures of peripheral thyroid hormone effects in thyrotoxic patients. Acta Endocrinol. (Copenh) 1985, 108, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Gereben, B.; Goncalves, C.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. Selective proteolysis of human type 2 deiodinase: A novel ubiquitin-proteasomal mediated mechanism for regulation of hormone activation. Mol. Endocrinol. 2000, 14, 1697–1708. [Google Scholar] [CrossRef]
- Sánchez, E.; Vargas, M.A.; Singru, P.S.; Pascual, I.; Romero, F.; Fekete, C.; Charli, J.L.; Lechan, R.M. Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology 2009, 150, 2283–2291. [Google Scholar] [CrossRef]
- Freitas, B.C.; Gereben, B.; Castillo, M.; Kalló, I.; Zeöld, A.; Egri, P.; Liposits, Z.; Zavacki, A.M.; Maciel, R.M.; Jo, S.; et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J. Clin. Investig. 2010, 120, 2206–2217. [Google Scholar] [CrossRef]
- Bocco, B.M.; Werneck-de-Castro, J.P.; Oliveira, K.C.; Fernandes, G.W.; Fonseca, T.L.; Nascimento, B.P.; McAninch, E.A.; Ricci, E.; Kvárta-Papp, Z.; Fekete, C.; et al. Type 2 Deiodinase Disruption in Astrocytes Results in Anxiety-Depressive-Like Behavior in Male Mice. Endocrinology 2016, 157, 3682–3695. [Google Scholar] [CrossRef]
- Fonseca, T.L.; Correa-Medina, M.; Campos, M.P.; Wittmann, G.; Werneck-de-Castro, J.P.; Arrojo e Drigo, R.; Mora-Garzon, M.; Ueta, C.B.; Caicedo, A.; Fekete, C.; et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J. Clin. Investig. 2013, 123, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Guadaño-Ferraz, A.; Obregon, M.J.; Germain, D.L.S.; Bernal, J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc. Natl. Acad. Sci. USA 1997, 94, 10391–10396. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Lazcano, I.; Sanchez-Jaramillo, E.; Uribe, R.M.; Jaimes-Hoy, L.; Joseph-Bravo, P.; Charli, J.L. Tanycytes and the Control of Thyrotropin-Releasing Hormone Flux into Portal Capillaries. Front. Endocrinol. 2019, 10, 401. [Google Scholar] [CrossRef]
- Schweizer, U.; Schlicker, C.; Braun, D.; Köhrle, J.; Steegborn, C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 10526–10531. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Salvatore, D. Deiodinases: The balance of thyroid hormone: Local impact of thyroid hormone inactivation. J. Endocrinol. 2011, 209, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Fliers, E.; Bianco, A.C.; Langouche, L.; Boelen, A. Thyroid function in critically ill patients. Lancet Diabetes Endocrinol. 2015, 3, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Falls, J.G.; Pulford, D.J.; Wylie, A.A.; Jirtle, R.L. Genomic imprinting: Implications for human disease. Am. J. Pathol. 1999, 154, 635–647. [Google Scholar] [CrossRef]
- Ohta, T.; Gray, T.A.; Rogan, P.K.; Buiting, K.; Gabriel, J.M.; Saitoh, S.; Muralidhar, B.; Bilienska, B.; Krajewska-Walasek, M.; Driscoll, D.J.; et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 1999, 64, 397–413. [Google Scholar] [CrossRef]
- Maher, E.R.; Reik, W. Beckwith-Wiedemann syndrome: Imprinting in clusters revisited. J. Clin. Investig. 2000, 105, 247–252.+27. [Google Scholar] [CrossRef]
- Hernandez, A.; Fiering, S.; Martinez, E.; Galton, V.A.; St Germain, D. The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology 2002, 143, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E.; Charalambous, M.; Saferali, A.; Fiering, S.; Naumova, A.K.; St Germain, D.; Ferguson-Smith, A.C.; Hernandez, A. Genomic imprinting variations in the mouse type 3 deiodinase gene between tissues and brain regions. Mol. Endocrinol. 2014, 28, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.J.; Towle, H.C.; Young, W.S., 3rd. Spatial and temporal expression of alpha- and beta-thyroid hormone receptor mRNAs, including the beta 2-subtype, in the developing mammalian nervous system. J. Neurosci. 1992, 12, 2288–2302. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Paterniti, I.; Campolo, M.; Di Paola, R.; Cuzzocrea, S.; Esposito, E. Exogenous T3 administration provides neuroprotection in a murine model of traumatic brain injury. Pharmacol. Res. 2013, 70, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Donangelo, I.; Abe, K.; Scremin, O.; Ke, S.; Li, F.; Milanesi, A.; Liu, Y.Y.; Brent, G.A. Thyroid hormone treatment activates protective pathways in both in vivo and in vitro models of neuronal injury. Mol. Cell. Endocrinol. 2017, 452, 120–130. [Google Scholar] [CrossRef]
- Zhang, L.; Cooper-Kuhn, C.M.; Nannmark, U.; Blomgren, K.; Kuhn, H.G. Stimulatory effects of thyroid hormone on brain angiogenesis in vivo and in vitro. J Cereb. Blood Flow Metab. 2010, 30, 323–335. [Google Scholar] [CrossRef]
- Cernak, I. Animal models of head trauma. NeuroRX 2005, 2, 410–422. [Google Scholar] [CrossRef]
- Potts, M.B.; Adwanikar, H.; Noble-Haeusslein, L.J. Models of traumatic cerebellar injury. Cerebellum 2009, 8, 211–221. [Google Scholar] [CrossRef]
- McAllister, T.W. Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 2011, 13, 287–300. [Google Scholar] [CrossRef]
- Dash, P.K.; Mach, S.A.; Moore, A.N. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J. Neurosci. Res. 2001, 63, 313–319. [Google Scholar] [CrossRef]
- Urrea, C.; Castellanos, D.A.; Sagen, J.; Tsoulfas, P.; Bramlett, H.M.; Dietrich, W.D. Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restor. Neurol. Neurosci. 2007, 25, 65–76. [Google Scholar]
- Sadana, P.; Coughlin, L.; Burke, J.; Woods, R.; Mdzinarishvili, A. Anti-edema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J. Neurol. Sci. 2015, 354, 37–45. [Google Scholar] [CrossRef]
- Merry, D.E.; Korsmeyer, S.J. Bcl-2 gene family in the nervous system. Annu. Rev. Neurosci. 1997, 20, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y.A. Caspases: Enemies within. Science 1998, 281, 312–1316. [Google Scholar] [CrossRef] [PubMed]
- Bathla, M.; Singh, M.; Relan, P. Prevalence of anxiety and depressive symptoms among patients with hypothyroidism. Indian J. Endocrinol. Metab. 2016, 20, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Khaleghzadeh-Ahangar, H.; Talebi, A.; Mohseni-Moghaddam, P. Thyroid Disorders and Development of Cognitive Impairment: A Review Study. Neuroendocrinology 2022, 112, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Venero, C.; Guadaño-Ferraz, A.; Herrero, A.I.; Nordström, K.; Manzano, J.; de Escobar, G.M.; Bernal, J.; Vennström, B. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev. 2005, 19, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Wallis, K.; Sjögren, M.; van Hogerlinden, M.; Silberberg, G.; Fisahn, A.; Nordström, K.; Larsson, L.; Westerblad, H.; Morreale de Escobar, G.; Shupliakov, O.; et al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J. Neurosci. 2008, 28, 1904–1915. [Google Scholar] [CrossRef]
- Chaalal, A.; Poirier, R.; Blum, D.; Laroche, S.; Enderlin, V. Thyroid Hormone Supplementation Restores Spatial Memory, Hippocampal Markers of Neuroinflammation, Plasticity-Related Signaling Molecules, and β-Amyloid Peptide Load in Hypothyroid Rats. Mol. Neurobiol. 2019, 56, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, V.C.; Araujo, S.M.; Pinheiro, F.C.; Poetini, M.R.; de Paula, M.T.; Meichtry, L.B.; de Almeida, F.P.; Musachio, E.A.S.; Guerra, G.P.; Prigol, M. Modulation of glutamate levels and Na+,K+-ATPase activity contributes to the chrysin memory recovery in hypothyroidism mice. Physiol. Behav. 2020, 222, 112892. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Evans, A.T.; Costall, B.; Smythe, J.W. Thyroid hormones, brain function and cognition: A brief review. Neurosci. Biobehav. Rev. 2002, 26, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Gerges, N.Z.; Aleisa, A.M.; Alkadhi, K.A. Levothyroxin restores hypothyroidism-induced impairment of hippocampus-dependent learning and memory: Behavioral, electrophysiological, and molecular studies. Hippocampus 2009, 19, 66–78. [Google Scholar] [CrossRef] [PubMed]
- de-Miranda, A.S.; Kuriyama, S.N.; da-Silva, C.S.; do-Nascimento, M.S.; Parente, T.E.; Paumgartten, F.J. Thyroid hormone disruption and cognitive impairment in rats exposed to PBDE during postnatal development. Reprod. Toxicol. 2016, 63, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.E.; Sanchez-Huerta, K.; Wood, C. Mild Thyroid Hormone Insufficiency During Development Compromises Activity-Dependent Neuroplasticity in the Hippocampus of Adult Male Rats. Endocrinology 2016, 157, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, S.M.; Vafaei, A.A.; Rashidy-Pour, A. Effects of maternal hypothyroidism during pregnancy on learning, memory and hippocampal BDNF in rat pups: Beneficial effects of exercise. Neuroscience 2016, 329, 151–161. [Google Scholar] [CrossRef]
- Shin, M.S.; Ko, I.G.; Kim, S.E.; Kim, B.K.; Kim, T.S.; Lee, S.H.; Hwang, D.S.; Kim, C.J.; Park, J.K.; Lim, B.V. Treadmill exercise ameliorates symptoms of methimazole-induced hypothyroidism through enhancing neurogenesis and suppressing apoptosis in the hippocampus of rat pups. Int. J. Dev. Neurosci. 2013, 31, 214–223. [Google Scholar] [CrossRef]
- Tong, H.; Chen, G.H.; Liu, R.Y.; Zhou, J.N. Age-related learning and memory impairments in adult-onset hypothyroidism in Kunming mice. Physiol. Behav. 2007, 91, 290–298. [Google Scholar] [CrossRef]
- Axelstad, M.; Hansen, P.R.; Boberg, J.; Bonnichsen, M.; Nellemann, C.; Lund, S.P.; Hougaard, K.S.; Hass, U. Developmental neurotoxicity of propylthiouracil (PTU) in rats: Relationship between transient hypothyroxinemia during development and long-lasting behavioural and functional changes. Toxicol. Appl. Pharmacol. 2008, 232, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, C.G.; Constantinou, C.; Giannakopoulou, D.; Giompres, P.; Margarity, M. Effect of adult onset hypothyroidism on behavioral parameters and acetylcholinesterase isoforms activity in specific brain regions of male mice. Physiol. Behav. 2016, 164, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Wilcoxon, J.S.; Nadolski, G.J.; Samarut, J.; Chassande, O.; Redei, E.E. Behavioral inhibition and impaired spatial learning and memory in hypothyroid mice lacking thyroid hormone receptor alpha. Behav. Brain. Res. 2007, 177, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Montero-Pedrazuela, A.; Fernández-Lamo, I.; Alieva, M.; Pereda-Pérez, I.; Venero, C.; Guadaño-Ferraz, A. Adult-onset hypothyroidism enhances fear memory and upregulates mineralocorticoid and glucocorticoid receptors in the amygdala. PLoS ONE 2011, 6, e26582. [Google Scholar] [CrossRef] [PubMed]
- Buras, A.; Battle, L.; Landers, E.; Nguyen, T.; Vasudevan, N. Thyroid hormones regulate anxiety in the male mouse. Horm. Behav. 2014, 65, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Hipólito, L.T.M.; Batista, T.H.; Dos Anjos-Garcia, T.; Giusti-Paiva, A.; Vilela, F.C. Methimazole-induced gestational hypothyroidism affects the offspring development and differently impairs the conditioned fear in male and female adulthood rodents. Int. J. Dev. Neurosci. 2023, 83, 108–120. [Google Scholar] [CrossRef]
- Melancia, F.; Servadio, M.; Schiavi, S.; Campolongo, P.; Giusti-Paiva, A.; Trezza, V. Testing the correlation between experimentally-induced hypothyroidism during pregnancy and autistic-like symptoms in the rat offspring. Behav. Brain Res. 2017, 321, 113–122. [Google Scholar] [CrossRef]
- Menezes, E.C.; Santos, P.R.; Goes, T.C.; Carvalho, V.C.B.; Teixeira-Silva, F.; Stevens, H.E.; Badauê-Passos, D.J.J. Effects of a rat model of gestational hypothyroidism on forebrain dopaminergic, GABAergic, and serotonergic systems and related behaviors. Behav. Brain Res. 2019, 2, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Alkadhi, K.A. Levothyroxin replacement therapy restores hypothyroidism induced impairment of L-LTP induction: Critical role of CREB. Brain Res. Bull. 2014, 100, 29–37. [Google Scholar] [CrossRef]
- Ambrogini, P.; Cuppini, R.; Ferri, P.; Mancini, C.; Ciaroni, S.; Voci, A.; Gerdoni, E.; Gallo, G. Thyroid hormones affect neurogenesis in the dentate gyrus of adult rat. Neuroendocrinology 2005, 81, 244–253. [Google Scholar] [CrossRef]
- Chaalal, A.; Poirier, R.; Blum, D.; Gillet, B.; Le Blanc, P.; Basquin, M.; Buee, L.; Laroche, S.; Enderlin, V. PTU-induced hypothyroidism in rats leads to several early neuropathological signs of Alzheimer’s disease in the hippocampus and spatial memory impairments. Hippocampus 2014, 24, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- O’Barr, S.A.; Oh, J.S.; Ma, C.; Brent, G.A.; Schultz, J.J. Thyroid hormone regulates endogenous amyloid-beta precursor protein gene expression and processing in both in vitro and in vivo models. Thyroid 2006, 16, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Belandia, B.; Latasa, M.J.; Villa, A.; Pascual, A. Thyroid hormone negatively regulates the transcriptional activity of the beta-amyloid precursor protein gene. J. Biol. Chem. 1998, 273, 30366–30371. [Google Scholar] [CrossRef]
- Villa, A.; Santiago, J.; Belandia, B.; Pascual, A. A response unit in the first exon of the beta-amyloid precursor protein gene containing thyroid hormone receptor and Sp1 binding sites mediates negative regulation by 3,5,30-triiodothyronine. Mol. Endocrinol. 2004, 18, 863–873. [Google Scholar] [CrossRef]
- Contreras-Jurado, C.; Pascual, A. Thyroid hormone regulation of APP (β-amyloid precursor protein) gene expression in brain and brain cultured cells. Neurochem. Int. 2012, 60, 484–487. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabatino, L.; Lapi, D.; Del Seppia, C. Factors and Mechanisms of Thyroid Hormone Activity in the Brain: Possible Role in Recovery and Protection. Biomolecules 2024, 14, 198. https://doi.org/10.3390/biom14020198
Sabatino L, Lapi D, Del Seppia C. Factors and Mechanisms of Thyroid Hormone Activity in the Brain: Possible Role in Recovery and Protection. Biomolecules. 2024; 14(2):198. https://doi.org/10.3390/biom14020198
Chicago/Turabian StyleSabatino, Laura, Dominga Lapi, and Cristina Del Seppia. 2024. "Factors and Mechanisms of Thyroid Hormone Activity in the Brain: Possible Role in Recovery and Protection" Biomolecules 14, no. 2: 198. https://doi.org/10.3390/biom14020198
APA StyleSabatino, L., Lapi, D., & Del Seppia, C. (2024). Factors and Mechanisms of Thyroid Hormone Activity in the Brain: Possible Role in Recovery and Protection. Biomolecules, 14(2), 198. https://doi.org/10.3390/biom14020198