
CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture Conditions of P. anserina
2.2. Metabolic Profiling of P. anserina
2.3. Global Proteome Profile of P. anserina
2.4. Mouse Cerebellar Metabolome
2.5. Mouse Metabolic Validation Study
2.6. Quantitative Immunoblots
2.7. Statistical Analyses
3. Results
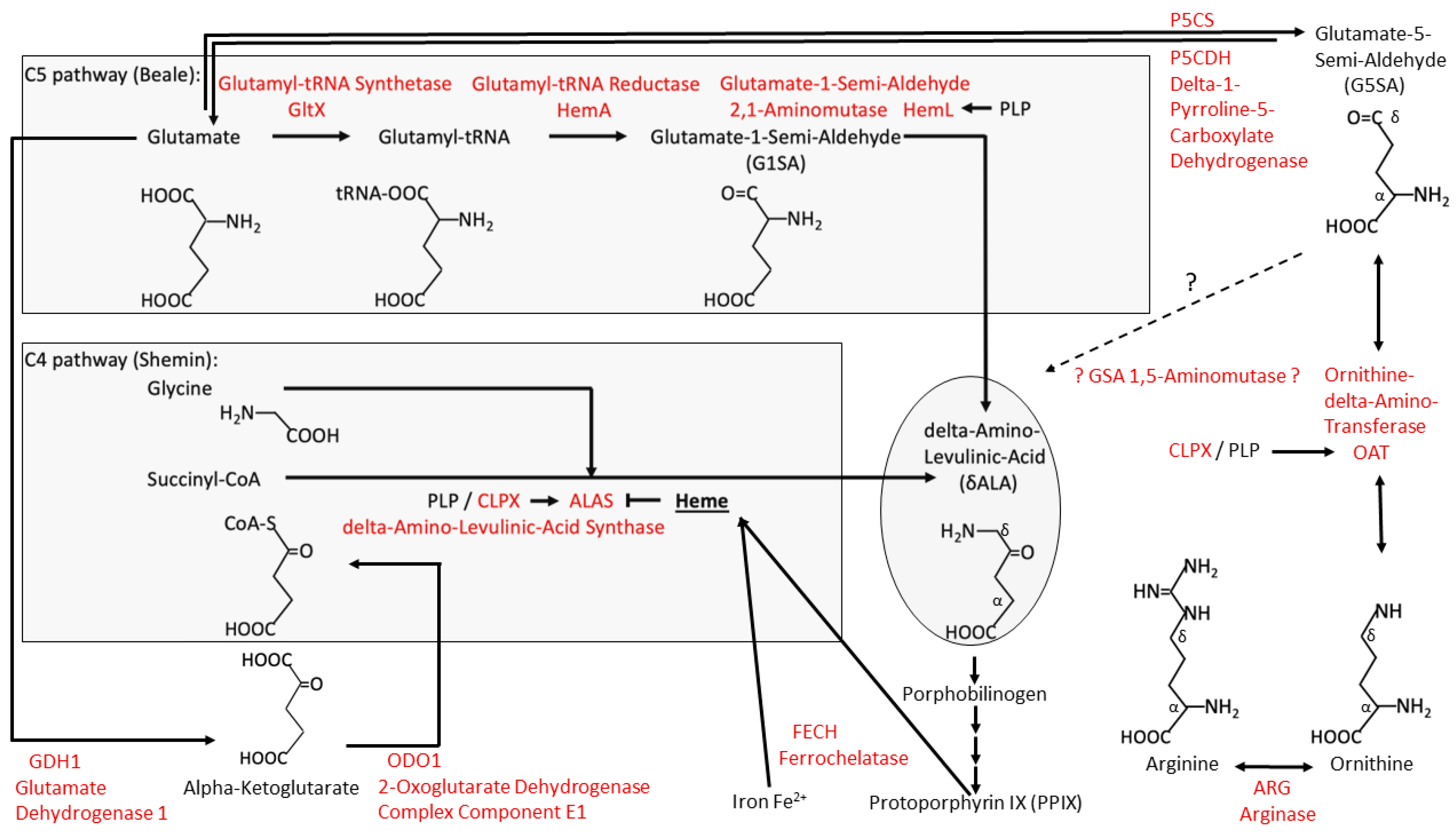
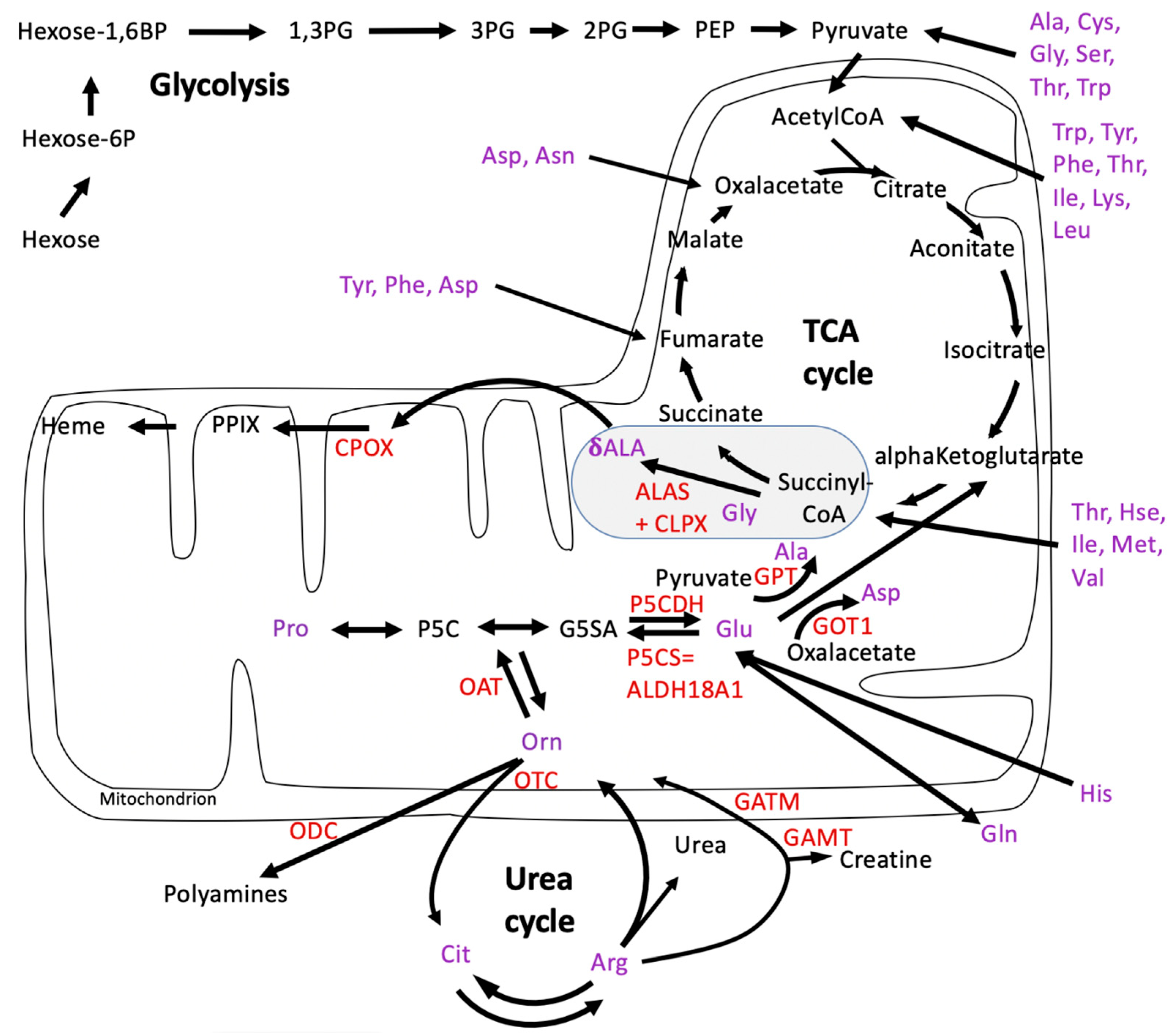
3.1. Re-Analysis of CLPP Mutation Effects on the P. anserina Metabolome Reveals CLPX as Only CLPP Substrate Associated with PLP, and Highlights OAT among the CLPP Interactors That Could Relate to Prominent Changes in Arginine and Ornithine Levels
3.2. Serine and Arginine Show the Strongest Reductions among Cognate Amino Acid Levels in CLPP-Null Mouse Cerebellum
3.3. Confirmatory Survey with Selected Non-Cognate Amino Acids in CLPP-Null Mouse Cerebellum
4. Discussion
4.1. Absence of CLPP Affects Amino Acid Metabsolism
4.2. Absence of CLPP Affects Mitoribosomal LSU and Its rRNA/tRNAVal/Phe
4.3. Absence of CLPP Affects SAM-Dependent Methyltransferases
4.4. Absence of CLPP Affects Molecular Chaperones and the UPRmt
4.5. Absence of CLPP Affects the Respiratory Chain
5. Conclusions
- (1)
- The consequent accumulation of CLPX with its PLP cofactor not only activates δALA production with downstream heme biosynthesis, but, in parallel, it also reduces the levels of other delta-amino acids such as Arg, Cit, and His. The consistent accumulation of ornithine delta-aminotransferase (OAT) and its probable activation by CLPX-PLP likely contribute to this effect. The halved Arg levels probably explain the growth deficit of CLPP-null mammals, so Arg supplementation in the diet might rescue the short stature and low muscle mass of PRLTS3 patients.
- (2)
- The absence of CLPP with excess CLPX alters the proteins in the LSU central protuberance and L7/L12 stalk, which are key for the processing and integration of tRNAVal/Phe as well as the SSU interactions required for complete mitoribosomal assembly.
- (3)
- Within the mitochondrial protein aggregation pathway, HSP70 accumulation is prominent.
- (4)
- An assembly problem involving the respiratory chain complex IV heme/copper-binding subunits could explain the mouse and P. anserina observations.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2PG | 2-phosphoglycerate |
| 5S rRNA | the smallest rRNA within the eukaryotic ribosomal LSU |
| 60S subunit | eukaryotic ribosomal LSU with sedimentation at 60 Svedberg units |
| AAA+ | ATPases associated with various cellular activities |
| ABAT | 4-aminobutyrate aminotransferase |
| ABC | ammonium bicarbonate |
| ACTB | beta-actin protein |
| AGAT | also known as GATM, L-arginine/glycine amidinotransferase |
| AGC | automatic gain control |
| AGXT2 | alanine–glyoxylate aminotransferase 2 |
| ALA | also known as deltaALA and delta-aminolevulinic acid |
| Ala | alanine |
| ALAS | delta-aminolevulinic acid synthase, generic |
| ALAS1 | delta-aminolevulinic acid synthase 1, non-specific |
| ALAS2 | delta-aminolevulinic acid synthase 1, erythroid-specific |
| ALDH18A1 | aldehyde dehydrogenase 18 family member A1 |
| Arg | arginine |
| ASMT | acetylserotonin O-methyltransferase |
| Asn | asparagine |
| Asp | aspartate |
| ATF4 | activating transcription factor 4 |
| ATP | adenosine trisphosphate |
| BAG-domain | Bcl-2-associated athanogene domain |
| BCAT2 | branched-chain amino acid transaminase 2 |
| BCL2 | BCL2 apoptosis regulator |
| C1/4/5/6 | chain composed of 1/4/5/6 carbons |
| C57BL/6 | inbred substrain 6 generated by C.C. Little, from Abbie Lathrop’s mouse 57, with nearly black coat |
| CI | respiratory chain complex I |
| CIV | respiratory chain complex IV |
| Cit | citrulline |
| CLPA-E | caseinolytic mitoch. matrix peptidase chaperone subunit A-E |
| CLPP | caseinolytic mitochondrial matrix peptidase proteolytic subunit |
| CLPX | caseinolytic mitochondrial matrix peptidase chaperone subunit X |
| CM liquid medium | complete medium containing glucose monohydrate |
| Co2+ | elemental cobalt as divalent cation |
| CoA | coenzyme A |
| COX2 | mitochondrially encoded cytochrome C oxidase II |
| COX7A | cytochrome C oxidase subunit 7A1 |
| COXFA4 | cytochrome C oxidase subunit FA4, also known as NDUFA4 |
| CPOX | coproporphyrinogen oxidase |
| CPT | carnitine palmitoyltransferase 2 |
| CTP | cytidine trisphosphate |
| Cys | cysteine |
| DNA | desoxyribonucleic acid |
| DnaK | E. coli chaperone protein |
| DTT | dithiothreitol |
| EARS2 | glutamyl-tRNA synthetase 2, mitochondrial |
| eIF6 | eukaryotic translation initiation factor 6 |
| ETNPPL | ethanolamine-phosphate phospholyase |
| ESI | electrospray ionization |
| ESSS | =NDUFB11, NADH-ubiquinone oxidoreductase subunit B11 |
| FAD | flavin adenine dinucleotide |
| FC | fold change |
| FDR | false discovery rate |
| Fe2+ | ferrous iron = iron(II), elemental iron as divalent cation |
| FECH | ferrochelatase |
| FeS clusters | iron–sulfur clusters |
| G1SA | glutamate-1-semialdehyde, also known as GSA |
| G5SA | glutamate-5-semialdehyde |
| GABA | gamma-amino-butyric acid |
| GAMT | guanidinoacetate N-methyltransferase |
| GAR1 | Gar1 ribonucleoprotein homolog |
| GATC | glutaminyl-tRNA synthase subunit C, mitochondrial |
| GATM | glycine amidinotransferase |
| GDH1 | glutamate decarboxylase 1 |
| GFM1/2 | translation elongation factor G, mitochondrial, variant 1/2 |
| GH | growth hormone |
| GLDC | glycine decarboxylase |
| Gln | glutamine |
| GltX | glutamate-tRNA ligase |
| Glu | glutamate |
| Gly | glycine |
| GO-term | gene ontology term |
| GOT1/2 | glutamic-oxaloacetic transaminase 1/2 |
| GPT | glutamic-pyruvic transaminase |
| GSSG | glutathione disulfide |
| GTP | guanosine triphosphate |
| H/ACA | sequence motifs H box (consensus ANANNA) and ACA box (ACA) |
| HARS2 | histidine-tRNA ligase, mitochondrial |
| HemA | glutamyl-tRNA reductase |
| HemL | glutamate-1-semialdehyde 2,1-aminomutase |
| Hexose-1,6BP | hexose-1,6-bisphosphate |
| Hexose-6P | hexose-6-phosphate |
| His | histidine |
| Hsc70 | heat shock cognate 71 kDa protein |
| Hse | homoserine |
| HSP70 | heat shock protein family A (Hsp70) member 4 |
| i-AAA | mitochondrial intermembrane space AAA+ protease |
| Ile | isoleucine |
| kDa | kiloDalton (molecular weight unit) |
| L7/L12 stalk | stalk structure in the mitoribosomal LSU, with proteins 7/12 |
| LARS2 | leucyl-tRNA synthetase 2, mitochondrial |
| LC/MS | liquid chromatography/mass spectrometry |
| Leu | leucine |
| LMBRD1 | lysosomal cobalamin transport escort protein LMBR1 |
| LonP | Lon peptidase 1 homolog, mitochondrial |
| Lsm7 | like-SM domain-containing protein 7, cytosolic |
| LSU | mitoribosomal large subunit |
| LYRM6 | protein 6 with conserved tripeptide (LYR) motif |
| m3G | 2,2,7-trimethyl guanosine |
| MCX1 | yeast CLPX homolog |
| Met | methionine |
| Mg2+ | elemental magnesium as divalent cation |
| 1-MHis | 1-methylhistamine |
| MRM | multiple reaction mode |
| MRPP1 | mitochondrial ribonuclease P protein 1, also known as TRMT10C |
| MRPP3 | mitochondrial ribonuclease P protein 3, also known as PRORP |
| MS | mass spectrometry |
| MRPL30 | large ribosomal subunit protein uL30m |
| MRPL33 | large ribosomal subunit protein bL33m |
| MTBE | methyl-tert-butyl ester |
| mt-CO3 | mitochondrially encoded cytochrome C oxidase III |
| mtDNA | mitochondrial DNA, nucleoid |
| 5,10-MTHF | 5,10-methylenetetrahydrofolate |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, methenyltetrahydrofolate cyclohydrolase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NDUFA1 | NADH:ubiquinone oxidoreductase subunit A1 |
| NDUFA2 | NADH:ubiquinone oxidoreductase subunit A2 |
| NDUFA2 | NADH:ubiquinone oxidoreductase subunit A3 |
| NDUFA4 | NADH:ubiquinone oxidoreductase subunit A4 |
| NDUFA6 | NADH:ubiquinone oxidoreductase subunit A6 |
| NDUFB2 | NADH:ubiquinone oxidoreductase subunit B2 |
| NDUFB11 | NADH:ubiquinone oxidoreductase subunit B11 |
| NDUFS6 | NADH:ubiquinone oxidoreductase subunit S6 |
| NFS1 | nitrogen fixing bacteria S-like protein 1, cysteine desulfurase |
| NOP10 | homolog of yeast Nop10p |
| NTMT1/2 | N-terminal Xaa-Pro-Lys N-methyltransferase 1/2 |
| OAT | ornithine aminotransferase |
| ODC | also known as ODC1, ornithine decarboxylase 1 |
| ODO1 | 2-oxoglutarate dehydrogenase complex component E1 |
| ONC201 | =dordaviprone: 11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo [7.4.0.02,6]trideca-1(9),5-dien-8-one |
| Orn | ornithine |
| OTC | ornithine transcarbamylase |
| P5C | delta-1-pyrroline-5-carboxylate |
| P5CDH | pyrroline-5-carboxylate dehydrogenase |
| P5CS | pyrroline-5-carboxylate synthetase, also known as ALDH18A1 |
| P5P | pyridoxal-5′-phosphate |
| Pa | Podospora anserina fungus |
| PADI | peptidylarginine deiminases |
| PAGE | polyacrylamide gel electrophoresis |
| PaIap | =B2B020 in UniProt; mitoch. intermembrane space AAA+ protease |
| PaSnf1 | =B2B4C1 in UniProt; sucrose non-fermenting complex, catalytic 1 |
| PEP | phosphoenolpyruvate |
| 1,3PG | 1,3-bisphospho-glycerate |
| 2PG | 2-phosphoglycerate |
| 3PG | 3-phosphoglycerate |
| PHD | prolyl-3-hydroxlase domain |
| Phe | phenylalanine |
| PHYKPL | 5-phosphohydroxy-L-lysine phospholyase |
| PLP | pyridoxal-5′-phosphate |
| PodAns | Podospora anserina fungus |
| PP/PE | polypropylene/polyethylene |
| PPIX | protoporphyrinogen IX |
| PRLTS3 | Perrault syndrome type 3 |
| Pro | proline |
| PRORP | protein-only RNase P catalytic subunit |
| RMND1 | required for meiotic nuclear division 1 homolog |
| RNA | ribonucleic acid |
| rRNA | ribosomal RNA |
| SAM | S-adenosyl methionine |
| SAM-MTases | S-adenosyl methionine-dependent methyltransferases |
| SAP domain | DNA-binding 35-residue motif, named after SAF-A/B, acinus, andPIAS, three proteins known to contain it |
| SDS | sodium dodecyl sulfate |
| Ser | serine |
| SHMT2 | serine hydroxymethyltransferase 2, mitochondrial |
| Sm domain | occurs in Sm proteins, named in honor of patient Stephanie Smith |
| SmG-like domain | spliceosomal core protein SmG, binds to AU dinucleotide |
| snoRNA | small nucleolar RNA |
| snRNA | small nuclear RNA |
| snRNP | small nuclear ribonucleoprotein |
| SSU | ribosomal small subunit |
| STRING | search tool for the retrieval of interacting genes/proteins |
| Tae1 | alpha N-terminal protein methyltransferase 1 |
| TCA cycle | tricarboxylic acid cycle |
| Thr | threonine |
| TRMT10C | tRNA methyltransferase 10C, mitochondrial RNase P subunit |
| tRNA | transfer RNA |
| tRNAVal/Phe | transfer RNA for valine or phenylalanine |
| Trp | tryptophan |
| TWNK | twinkle |
| Tyr | tyrosine |
| UniProt | public database about proteins, unifies nomenclature |
| UniProt-ID | UniProt protein identifier number |
| UPRmt | mitochondrial unfolded protein response |
| UTP | uridine triphosphate |
| v/v | volume per volume |
| WT | Wild-type |
| ZFE | Zentrale Forschungs-Einrichtung |
References
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 2016, 14, 33–44. [Google Scholar] [CrossRef]
- Aubin-Tam, M.E.; Olivares, A.O.; Sauer, R.T.; Baker, T.A.; Lang, M.J. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 2011, 145, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife 2020, 9, e52774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hartling, J.A.; Flanagan, J.M. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 1997, 91, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Petereit, J.; Millar, A.H. Loss of conserved mitochondrial CLPP and its functions lead to different phenotypes in plants and other organisms. Plant Signal. Behav. 2020, 15, 1831789. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11, 2370. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Bhandari, V.; Houry, W.A. Substrates and interactors of the ClpP protease in the mitochondria. Curr. Opin. Chem. Biol. 2022, 66, 102078. [Google Scholar] [CrossRef]
- Key, J.; Kohli, A.; Barcena, C.; Lopez-Otin, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1(+/−) Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef]
- Chandu, D.; Nandi, D. Comparative genomics and functional roles of the ATP-dependent proteases Lon and Clp during cytosolic protein degradation. Res. Microbiol. 2004, 155, 710–719. [Google Scholar] [CrossRef]
- Maurizi, M.R.; Clark, W.P.; Katayama, Y.; Rudikoff, S.; Pumphrey, J.; Bowers, B.; Gottesman, S. Sequence and structure of Clp P, the proteolytic component of the ATP-dependent Clp protease of Escherichia coli. J. Biol. Chem. 1990, 265, 12536–12545. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, L.; Dembowski, M.; Neupert, W.; Langer, T. Mcx1p, a ClpX homologue in mitochondria of Saccharomyces cerevisiae. FEBS Lett. 1998, 438, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 2015, 161, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B(12), folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- van der Vorm, L.N.; Paw, B.H. Studying disorders of vertebrate iron and heme metabolism using zebrafish. Methods Cell Biol. 2017, 138, 193–220. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortes, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030–2033. [Google Scholar] [CrossRef]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef]
- Shemin, D. An illustration of the use of isotopes: The biosynthesis of porphyrins. Bioessays 1989, 10, 30–35. [Google Scholar] [CrossRef]
- Beale, S.I.; Castelfranco, P.A. The Biosynthesis of delta-Aminolevulinic Acid in Higher Plants: II. Formation of C-delta-Aminolevulinic Acid from Labeled Precursors in Greening Plant Tissues. Plant Physiol. 1974, 53, 297–303. [Google Scholar] [CrossRef]
- Iida, K.; Mimura, I.; Kajiwara, M. Evaluation of two biosynthetic pathways to delta-aminolevulinic acid in Euglena gracilis. Eur. J. Biochem. 2002, 269, 291–297. [Google Scholar] [CrossRef]
- Petricek, M.; Petrickova, K.; Havlicek, L.; Felsberg, J. Occurrence of two 5-aminolevulinate biosynthetic pathways in Streptomyces nodosus subsp. asukaensis is linked with the production of asukamycin. J. Bacteriol. 2006, 188, 5113–5123. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Key, J.; Gispert, S.; Koepf, G.; Steinhoff-Wagner, J.; Reichlmeir, M.; Auburger, G. Translation fidelity and respiration deficits in CLPP-deficient tissues: Mechanistic insights from mitochondrial complexome. Int. J. Mol. Sci. 2023, 24, 17503. [Google Scholar] [CrossRef]
- Koper, K.; Han, S.W.; Pastor, D.C.; Yoshikuni, Y.; Maeda, H.A. Evolutionary origin and functional diversification of aminotransferases. J. Biol. Chem. 2022, 298, 102122. [Google Scholar] [CrossRef]
- Cellini, B.; Montioli, R.; Oppici, E.; Astegno, A.; Voltattorni, C.B. The chaperone role of the pyridoxal 5′-phosphate and its implications for rare diseases involving B6-dependent enzymes. Clin. Biochem. 2014, 47, 158–165. [Google Scholar] [CrossRef]
- Liang, J.; Han, Q.; Tan, Y.; Ding, H.; Li, J. Current Advances on Structure-Function Relationships of Pyridoxal 5′-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019, 6, 4. [Google Scholar] [CrossRef]
- Shin, B.S.; Katoh, T.; Gutierrez, E.; Kim, J.R.; Suga, H.; Dever, T.E. Amino acid substrates impose polyamine, eIF5A, or hypusine requirement for peptide synthesis. Nucleic Acids Res. 2017, 45, 8392–8402. [Google Scholar] [CrossRef]
- Pegg, A.E. Toxicity of polyamines and their metabolic products. Chem. Res. Toxicol. 2013, 26, 1782–1800. [Google Scholar] [CrossRef]
- di Salvo, M.L.; Budisa, N.; Contestabile, R. PLP-dependent Enzymes: A Powerful Tool for Metabolic Synthesis of Non-canonical Amino Acids. 2012. Available online: https://www.beilstein-institut.de/download/65/plp-dependent_enzymes_a_powerful_tool_for_metabolic_synthesis_of_non-canonical_amino_acids_.pdf (accessed on 6 February 2024).
- Obermaier, S.; Muller, M. Ibotenic Acid Biosynthesis in the Fly Agaric Is Initiated by Glutamate Hydroxylation. Angew. Chem. Int. Ed. Engl. 2020, 59, 12432–12435. [Google Scholar] [CrossRef]
- Chen, M.; Liu, C.T.; Tang, Y. Discovery and Biocatalytic Application of a PLP-Dependent Amino Acid gamma-Substitution Enzyme That Catalyzes C-C Bond Formation. J. Am. Chem. Soc. 2020, 142, 10506–10515. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef]
- Zhou, Z.; Fan, Y.; Zong, R.; Tan, K. The mitochondrial unfolded protein response: A multitasking giant in the fight against human diseases. Ageing Res. Rev. 2022, 81, 101702. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef]
- Levchenko, I.; Seidel, M.; Sauer, R.T.; Baker, T.A. A specificity-enhancing factor for the ClpXP degradation machine. Science 2000, 289, 2354–2356. [Google Scholar] [CrossRef] [PubMed]
- Lytvynenko, I.; Paternoga, H.; Thrun, A.; Balke, A.; Muller, T.A.; Chiang, C.H.; Nagler, K.; Tsaprailis, G.; Anders, S.; Bischofs, I.; et al. Alanine Tails Signal Proteolysis in Bacterial Ribosome-Associated Quality Control. Cell 2019, 178, 76–90.e22. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Tantray, I.; Lim, J.; Chen, S.; Li, Y.; Davis, Z.; Sitron, C.; Dong, J.; Gispert, S.; Auburger, G.; et al. MISTERMINATE Mechanistically Links Mitochondrial Dysfunction with Proteostasis Failure. Mol. Cell 2019, 75, 835–848.e838. [Google Scholar] [CrossRef]
- Huter, P.; Arenz, S.; Bock, L.V.; Graf, M.; Frister, J.O.; Heuer, A.; Peil, L.; Starosta, A.L.; Wohlgemuth, I.; Peske, F.; et al. Structural Basis for Polyproline-Mediated Ribosome Stalling and Rescue by the Translation Elongation Factor EF-P. Mol. Cell 2017, 68, 515–527.e516. [Google Scholar] [CrossRef]
- Konovalova, S.; Hilander, T.; Loayza-Puch, F.; Rooijers, K.; Agami, R.; Tyynismaa, H. Exposure to arginine analog canavanine induces aberrant mitochondrial translation products, mitoribosome stalling, and instability of the mitochondrial proteome. Int. J. Biochem. Cell Biol. 2015, 65, 268–274. [Google Scholar] [CrossRef]
- Ou, X.; Cao, J.; Cheng, A.; Peppelenbosch, M.P.; Pan, Q. Errors in translational decoding: tRNA wobbling or misincorporation? PLoS Genet. 2019, 15, e1008017. [Google Scholar] [CrossRef]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 12862. [Google Scholar] [CrossRef]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Theunissen, T.E.; Szklarczyk, R.; Gerards, M.; Hellebrekers, D.M.; Mulder-Den Hartog, E.N.; Vanoevelen, J.; Kamps, R.; de Koning, B.; Rutledge, S.L.; Schmitt-Mechelke, T.; et al. Specific MRI Abnormalities Reveal Severe Perrault Syndrome due to CLPP Defects. Front. Neurol. 2016, 7, 203. [Google Scholar] [CrossRef]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10, 3354. [Google Scholar] [CrossRef]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2022, 141, 805–819. [Google Scholar] [CrossRef]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e729. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, P.; Wei, B.; Chen, L.; Song, X.; Pan, Y.; Li, J.; Gan, J.; Zhang, T.; Yang, C.G. Assessment of the structure-activity relationship and antileukemic activity of diacylpyramide compounds as human ClpP agonists. Eur. J. Med. Chem. 2023, 258, 115577. [Google Scholar] [CrossRef]
- Key, J.; Gispert, S.; Koornneef, L.; Sleddens-Linkels, E.; Kohli, A.; Torres-Odio, S.; Koepf, G.; Amr, S.; Reichlmeir, M.; Harter, P.N.; et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells 2022, 12, 52. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Becker, C.; Kukat, A.; Szczepanowska, K.; Hermans, S.; Senft, K.; Brandscheid, C.P.; Maiti, P.; Trifunovic, A. CLPP deficiency protects against metabolic syndrome but hinders adaptive thermogenesis. EMBO Rep. 2018, 19, e45126. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef]
- Key, J.; Maletzko, A.; Kohli, A.; Gispert, S.; Torres-Odio, S.; Wittig, I.; Heidler, J.; Barcena, C.; Lopez-Otin, C.; Lei, Y.; et al. Loss of mitochondrial ClpP, Lonp1, and Tfam triggers transcriptional induction of Rnf213, a susceptibility factor for moyamoya disease. Neurogenetics 2020, 21, 187–203. [Google Scholar] [CrossRef]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of Mitochondrial Protease CLPP Activates Type I IFN Responses through the Mitochondrial DNA-cGAS-STING Signaling Axis. J. Immunol. 2021, 206, 1890–1900. [Google Scholar] [CrossRef]
- Maletzko, A.; Key, J.; Wittig, I.; Gispert, S.; Koepf, G.; Canet-Pons, J.; Torres-Odio, S.; West, A.P.; Auburger, G. Increased presence of nuclear DNAJA3 and upregulation of cytosolic STAT1 and of nucleic acid sensors trigger innate immunity in the ClpP-null mouse. Neurogenetics 2021, 22, 297–312. [Google Scholar] [CrossRef]
- Osiewacz, H.D.; Hamann, A.; Zintel, S. Assessing Organismal Aging in the Filamentous Fungus Podospora anserina. In Cell Senescence: Methods and Protocols, Methods in Molecular Biology; Galluzzi, L., Vitale, I., Kepp, O., Kroemer, G., Eds.; Humana: Totowa, NJ, USA, 2013; Volume 965. [Google Scholar]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef]
- Heinz, D.; Krotova, E.; Hamann, A.; Osiewacz, H.D. Simultaneous Ablation of the Catalytic AMPK alpha-Subunit SNF1 and Mitochondrial Matrix Protease CLPP Results in Pronounced Lifespan Extension. Front. Cell Dev. Biol. 2021, 9, 616520. [Google Scholar] [CrossRef]
- Fischer, F.; Langer, J.D.; Osiewacz, H.D. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Hohne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020, 11, 1643. [Google Scholar] [CrossRef] [PubMed]
- Petereit, J.; Duncan, O.; Murcha, M.W.; Fenske, R.; Cincu, E.; Cahn, J.; Pruzinska, A.; Ivanova, A.; Kollipara, L.; Wortelkamp, S.; et al. Mitochondrial CLPP2 Assists Coordination and Homeostasis of Respiratory Complexes. Plant Physiol. 2020, 184, 148–164. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Meierhofer, D.; Halbach, M.; Sen, N.E.; Gispert, S.; Auburger, G. Ataxin-2 (Atxn2)-Knock-Out Mice Show Branched Chain Amino Acids and Fatty Acids Pathway Alterations. Mol. Cell. Proteomics 2016, 15, 1728–1739. [Google Scholar] [CrossRef]
- Gielisch, I.; Meierhofer, D. Metabolome and proteome profiling of complex I deficiency induced by rotenone. J. Proteome Res. 2015, 14, 224–235. [Google Scholar] [CrossRef]
- Funck, D.; Stadelhofer, B.; Koch, W. Ornithine-delta-aminotransferase is essential for arginine catabolism but not for proline biosynthesis. BMC Plant Biol. 2008, 8, 40. [Google Scholar] [CrossRef]
- Du, J.; Zhu, S.; Lim, R.R.; Chao, J.R. Proline metabolism and transport in retinal health and disease. Amino Acids 2021, 53, 1789–1806. [Google Scholar] [CrossRef]
- Hofsetz, E.; Demir, F.; Szczepanowska, K.; Kukat, A.; Kizhakkedathu, J.N.; Trifunovic, A.; Huesgen, P.F. The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol. Cell. Proteom. 2020, 19, 1330–1345. [Google Scholar] [CrossRef]
- Monne, M.; Marobbio, C.M.T.; Agrimi, G.; Palmieri, L.; Palmieri, F. Mitochondrial transport and metabolism of the major methyl donor and versatile cofactor S-adenosylmethionine, and related diseases: A review (dagger). IUBMB Life 2022, 74, 573–591. [Google Scholar] [CrossRef]
- Rebelo-Guiomar, P.; Pellegrino, S.; Dent, K.C.; Sas-Chen, A.; Miller-Fleming, L.; Garone, C.; Van Haute, L.; Rogan, J.F.; Dinan, A.; Firth, A.E.; et al. A late-stage assembly checkpoint of the human mitochondrial ribosome large subunit. Nat. Commun. 2022, 13, 929. [Google Scholar] [CrossRef] [PubMed]
- Koditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef]
- Stranska, J.; Kopecny, D.; Tylichova, M.; Snegaroff, J.; Sebela, M. Ornithine delta-aminotransferase: An enzyme implicated in salt tolerance in higher plants. Plant Signal. Behav. 2008, 3, 929–935. [Google Scholar] [CrossRef]
- Llacer, J.L.; Fita, I.; Rubio, V. Arginine and nitrogen storage. Curr. Opin. Struct. Biol. 2008, 18, 673–681. [Google Scholar] [CrossRef]
- Pencharz, P.B. Assessment of protein nutritional status in children. Pediatr. Blood Cancer 2008, 50, 445–446, discussion 451. [Google Scholar] [CrossRef]
- Sodero, G.; Mariani, F.; Caprarelli, M.; Agazzi, C.; Quarta, L.; Benacquista, L.; Rigante, D.; Clelia, C. Growth hormone responses during arginine and clonidine stimulation test: Correlations with patients’ auxological and metabolic parameters in a single centre study. Growth Horm. IGF Res. 2023, 68, 101522. [Google Scholar] [CrossRef]
- Stefani, G.P.; Marmett, B.; Alves, J.P.; Moller, G.B.; Heck, T.G.; Frizzo, M.N.; Di Domenico, M.; Motta, G.A.; Dal Lago, P.; Nunes, R.B.; et al. Resistance training and L-arginine supplementation are determinant in genomic stability, cardiac contractility and muscle mass development in rats. PLoS ONE 2018, 13, e0204858. [Google Scholar] [CrossRef]
- Vanderniet, J.A.; Benitez-Aguirre, P.Z.; Broderick, C.R.; Kelley, R.I.; Balasubramaniam, S. Barth syndrome with severe dilated cardiomyopathy and growth hormone resistance: A case report. J. Pediatr. Endocrinol. Metab. 2021, 34, 951–955. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Brosnan, J.T. Histidine Metabolism and Function. J. Nutr. 2020, 150, 2570S–2575S. [Google Scholar] [CrossRef]
- Hoang, X.L.T.; Prerostova, S.; Thu, N.B.A.; Thao, N.P.; Vankova, R.; Tran, L.P. Histidine Kinases: Diverse Functions in Plant Development and Responses to Environmental Conditions. Annu. Rev. Plant Biol. 2021, 72, 297–323. [Google Scholar] [CrossRef]
- Imura, K.; Okada, A. Amino acid metabolism in pediatric patients. Nutrition 1998, 14, 143–148. [Google Scholar] [CrossRef]
- Mercer, L.P.; Dodds, S.J.; Weber, M.D.; Dunn, J.D. Histidine, histamine, and the neuroregulation of food intake: A review and hypothesis. Nutrition 1990, 6, 273–277. [Google Scholar]
- Moro, J.; Tome, D.; Schmidely, P.; Demersay, T.C.; Azzout-Marniche, D. Histidine: A Systematic Review on Metabolism and Physiological Effects in Human and Different Animal Species. Nutrients 2020, 12, 1414. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef]
- Hirasawa, N. Expression of Histidine Decarboxylase and Its Roles in Inflammation. Int. J. Mol. Sci. 2019, 20, 376. [Google Scholar] [CrossRef]
- Brown, A.; Amunts, A.; Bai, X.C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef]
- Wahl, M.C.; Moller, W. Structure and function of the acidic ribosomal stalk proteins. Curr. Protein Pept. Sci. 2002, 3, 93–106. [Google Scholar] [CrossRef]
- Rorbach, J.; Gao, F.; Powell, C.A.; D’Souza, A.; Lightowlers, R.N.; Minczuk, M.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial ribosomes can switch their structural RNA composition. Proc. Natl. Acad. Sci. USA 2016, 113, 12198–12201. [Google Scholar] [CrossRef]
- Miluzio, A.; Beugnet, A.; Volta, V.; Biffo, S. Eukaryotic initiation factor 6 mediates a continuum between 60S ribosome biogenesis and translation. EMBO Rep. 2009, 10, 459–465. [Google Scholar] [CrossRef]
- Gartmann, M.; Blau, M.; Armache, J.P.; Mielke, T.; Topf, M.; Beckmann, R. Mechanism of eIF6-mediated inhibition of ribosomal subunit joining. J. Biol. Chem. 2010, 285, 14848–14851. [Google Scholar] [CrossRef]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef]
- Jaako, P.; Faille, A.; Tan, S.; Wong, C.C.; Escudero-Urquijo, N.; Castro-Hartmann, P.; Wright, P.; Hilcenko, C.; Adams, D.J.; Warren, A.J. eIF6 rebinding dynamically couples ribosome maturation and translation. Nat. Commun. 2022, 13, 1562. [Google Scholar] [CrossRef]
- Pesce, E.; Miluzio, A.; Turcano, L.; Minici, C.; Cirino, D.; Calamita, P.; Manfrini, N.; Oliveto, S.; Ricciardi, S.; Grifantini, R.; et al. Discovery and Preliminary Characterization of Translational Modulators that Impair the Binding of eIF6 to 60S Ribosomal Subunits. Cells 2020, 9, 172. [Google Scholar] [CrossRef]
- Balogh, E.; Chandler, J.C.; Varga, M.; Tahoun, M.; Menyhard, D.K.; Schay, G.; Goncalves, T.; Hamar, R.; Legradi, R.; Szekeres, A.; et al. Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc. Natl. Acad. Sci. USA 2020, 117, 15137–15147. [Google Scholar] [CrossRef]
- De Zoysa, M.D.; Yu, Y.T. Posttranscriptional RNA Pseudouridylation. Enzymes 2017, 41, 151–167. [Google Scholar] [CrossRef]
- Mund, M.; Neu, A.; Ullmann, J.; Neu, U.; Sprangers, R. Structure of the LSm657 complex: An assembly intermediate of the LSm1-7 and LSm2-8 rings. J. Mol. Biol. 2011, 414, 165–176. [Google Scholar] [CrossRef]
- Jakubowski, H. Misacylation of tRNALys with noncognate amino acids by lysyl-tRNA synthetase. Biochemistry 1999, 38, 8088–8093. [Google Scholar] [CrossRef]
- Bauerle, M.R.; Schwalm, E.L.; Booker, S.J. Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem. 2015, 290, 3995–4002. [Google Scholar] [CrossRef]
- Ishiguro, K.; Arai, T.; Suzuki, T. Depletion of S-adenosylmethionine impacts on ribosome biogenesis through hypomodification of a single rRNA methylation. Nucleic Acids Res. 2019, 47, 4226–4239. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Xiao, H.; Bonanno, J.B.; Kalyanaraman, C.; Brown, S.; Tang, X.; Al-Obaidi, N.F.; Patskovsky, Y.; Babbitt, P.C.; Jacobson, M.P.; et al. Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature 2013, 498, 123–126. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ren, D.; Jeon, B.; Liu, H.W. S-Adenosylmethionine: More than just a methyl donor. Nat. Prod. Rep. 2023, 40, 1521–1549. [Google Scholar] [CrossRef]
- Wang, S.C. Cobalamin-dependent radical S-adenosyl-l-methionine enzymes in natural product biosynthesis. Nat. Prod. Rep. 2018, 35, 707–720. [Google Scholar] [CrossRef]
- Kishimoto, S.; Hara, K.; Hashimoto, H.; Hirayama, Y.; Champagne, P.A.; Houk, K.N.; Tang, Y.; Watanabe, K. Enzymatic one-step ring contraction for quinolone biosynthesis. Nat. Commun. 2018, 9, 2826. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Poree, F.H.; Gaslonde, T.; Lalucque, H.; Chapeland-Leclerc, F.; Ruprich-Robert, G. Functional characterization of the sterigmatocystin secondary metabolite gene cluster in the filamentous fungus Podospora anserina: Involvement in oxidative stress response, sexual development, pigmentation and interspecific competitions. Environ. Microbiol. 2019, 21, 3011–3026. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.J.; Dobson, A.D. Molecular biology of mycotoxin biosynthesis. FEMS Microbiol. Lett. 1999, 175, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Webb, K.J.; Lipson, R.S.; Al-Hadid, Q.; Whitelegge, J.P.; Clarke, S.G. Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry 2010, 49, 5225–5235. [Google Scholar] [CrossRef]
- Alamgir, M.; Eroukova, V.; Jessulat, M.; Xu, J.; Golshani, A. Chemical-genetic profile analysis in yeast suggests that a previously uncharacterized open reading frame, YBR261C, affects protein synthesis. BMC Genom. 2008, 9, 583. [Google Scholar] [CrossRef]
- Chen, P.; Paschoal Sobreira, T.J.; Hall, M.C.; Hazbun, T.R. Discovering the N-Terminal Methylome by Repurposing of Proteomic Datasets. J. Proteome Res. 2021, 20, 4231–4247. [Google Scholar] [CrossRef]
- Bhatta, A.; Dienemann, C.; Cramer, P.; Hillen, H.S. Structural basis of RNA processing by human mitochondrial RNase P. Nat. Struct. Mol. Biol. 2021, 28, 713–723. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Thompson, K.; Alston, C.L.; Morris, A.A.M.; He, L.; Assouline, Z.; Rio, M.; Bahi-Buisson, N.; Pyle, A.; Griffin, H.; et al. Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies. Am. J. Hum. Genet. 2016, 98, 993–1000. [Google Scholar] [CrossRef]
- Oerum, S.; Roovers, M.; Rambo, R.P.; Kopec, J.; Bailey, H.J.; Fitzpatrick, F.; Newman, J.A.; Newman, W.G.; Amberger, A.; Zschocke, J.; et al. Structural insight into the human mitochondrial tRNA purine N1-methyltransferase and ribonuclease P complexes. J. Biol. Chem. 2018, 293, 12862–12876. [Google Scholar] [CrossRef]
- Reinhard, L.; Sridhara, S.; Hallberg, B.M. The MRPP1/MRPP2 complex is a tRNA-maturation platform in human mitochondria. Nucleic Acids Res. 2017, 45, 12469–12480. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Jensen, O.N.; Mann, M.; Schagger, H.; Osiewacz, H.D. Identification and characterization of PaMTH1, a putative O-methyltransferase accumulating during senescence of Podospora anserina cultures. Curr. Genet. 2000, 37, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kunstmann, B.; Osiewacz, H.D. Over-expression of an S-adenosylmethionine-dependent methyltransferase leads to an extended lifespan of Podospora anserina without impairments in vital functions. Aging Cell 2008, 7, 651–662. [Google Scholar] [CrossRef]
- Chatterjee, D.; Kudlinzki, D.; Linhard, V.; Saxena, K.; Schieborr, U.; Gande, S.L.; Wurm, J.P.; Wohnert, J.; Abele, R.; Rogov, V.V.; et al. Structure and Biophysical Characterization of the S-Adenosylmethionine-dependent O-Methyltransferase PaMTH1, a Putative Enzyme Accumulating during Senescence of Podospora anserina. J. Biol. Chem. 2015, 290, 16415–16430. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Hofer, S.J.; Simon, A.K.; Bergmann, M.; Eisenberg, T.; Kroemer, G.; Madeo, F. Mechanisms of spermidine-induced autophagy and geroprotection. Nat. Aging 2022, 2, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Hofer, S.J.; Pendl, T.; Bauer, M.A.; Eisenberg, T.; Carmona-Gutierrez, D.; Kroemer, G. Nutritional Aspects of Spermidine. Annu. Rev. Nutr. 2020, 40, 135–159. [Google Scholar] [CrossRef]
- Sagar, N.A.; Tarafdar, S.; Agarwal, S.; Tarafdar, A.; Sharma, S. Polyamines: Functions, Metabolism, and Role in Human Disease Management. Med. Sci. 2021, 9, 44. [Google Scholar] [CrossRef]
- Singh, G.; Pandey, R.; Anthony, E.R.; Chandra, S.; Mehrotra, D. Expression and bioinformatics analyses show HSP70 complements BCL2 action in oral carcinogenesis. J. Oral Biol. Craniofacial Res. 2022, 12, 599–603. [Google Scholar] [CrossRef]
- Gailus, S.; Hohne, W.; Gasnier, B.; Nurnberg, P.; Fowler, B.; Rutsch, F. Insights into lysosomal cobalamin trafficking: Lessons learned from cblF disease. J. Mol. Med. 2010, 88, 459–466. [Google Scholar] [CrossRef]
- Parkhitko, A.A.; Jouandin, P.; Mohr, S.E.; Perrimon, N. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 2019, 18, e13034. [Google Scholar] [CrossRef]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef]
- Kadenbach, B. Regulation of Mammalian 13-Subunit Cytochrome c Oxidase and Binding of other Proteins: Role of NDUFA4. Trends Endocrinol. Metab. 2017, 28, 761–770. [Google Scholar] [CrossRef]
- Yagil, C.; Varadi-Levi, R.; Yagil, Y. A novel mutation in the NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4 (Ndufa4) gene links mitochondrial dysfunction to the development of diabetes in a rodent model. Dis. Model Mech. 2018, 11, dmm036699. [Google Scholar] [CrossRef]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef]
- Sorouri, M.; Chang, T.; Jesudhasan, P.; Pinkham, C.; Elde, N.C.; Hancks, D.C. Signatures of host-pathogen evolutionary conflict reveal MISTR-A conserved MItochondrial STress Response network. PLoS Biol. 2020, 18, e3001045. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Taanman, J.W. NDUFA4 (Renamed COXFA4) Is a Cytochrome-c Oxidase Subunit. Trends Endocrinol. Metab. 2018, 29, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Lopez-Calcerrada, S.; Sierra-Magro, A.; Perez-Perez, R.; Formosa, L.E.; Hock, D.H.; Illescas, M.; Penas, A.; Brischigliaro, M.; Ding, S.; et al. Two independent respiratory chains adapt OXPHOS performance to glycolytic switch. Cell Metab. 2022, 34, 1792–1808.e1796. [Google Scholar] [CrossRef]
- Sorge, S.; Theelke, J.; Yildirim, K.; Hertenstein, H.; McMullen, E.; Muller, S.; Altburger, C.; Schirmeier, S.; Lohmann, I. ATF4-Induced Warburg Metabolism Drives Over-Proliferation in Drosophila. Cell Rep. 2020, 31, 107659. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- Soto, I.C.; Fontanesi, F.; Liu, J.; Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 2012, 1817, 883–897. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-Lopez, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 43. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Gispert, S.; Kandi, A.R.; Heinz, D.; Hamann, A.; Osiewacz, H.D.; Meierhofer, D.; Auburger, G. CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation. Biomolecules 2024, 14, 241. https://doi.org/10.3390/biom14020241
Key J, Gispert S, Kandi AR, Heinz D, Hamann A, Osiewacz HD, Meierhofer D, Auburger G. CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation. Biomolecules. 2024; 14(2):241. https://doi.org/10.3390/biom14020241
Chicago/Turabian StyleKey, Jana, Suzana Gispert, Arvind Reddy Kandi, Daniela Heinz, Andrea Hamann, Heinz D. Osiewacz, David Meierhofer, and Georg Auburger. 2024. "CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation" Biomolecules 14, no. 2: 241. https://doi.org/10.3390/biom14020241
APA StyleKey, J., Gispert, S., Kandi, A. R., Heinz, D., Hamann, A., Osiewacz, H. D., Meierhofer, D., & Auburger, G. (2024). CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation. Biomolecules, 14(2), 241. https://doi.org/10.3390/biom14020241