MicroRNA Expression in Cystic Fibrosis Airway Epithelium


{kind=link}
Abstract
:1. Introduction
1.1. Cystic Fibrosis
1.2. MicroRNAs
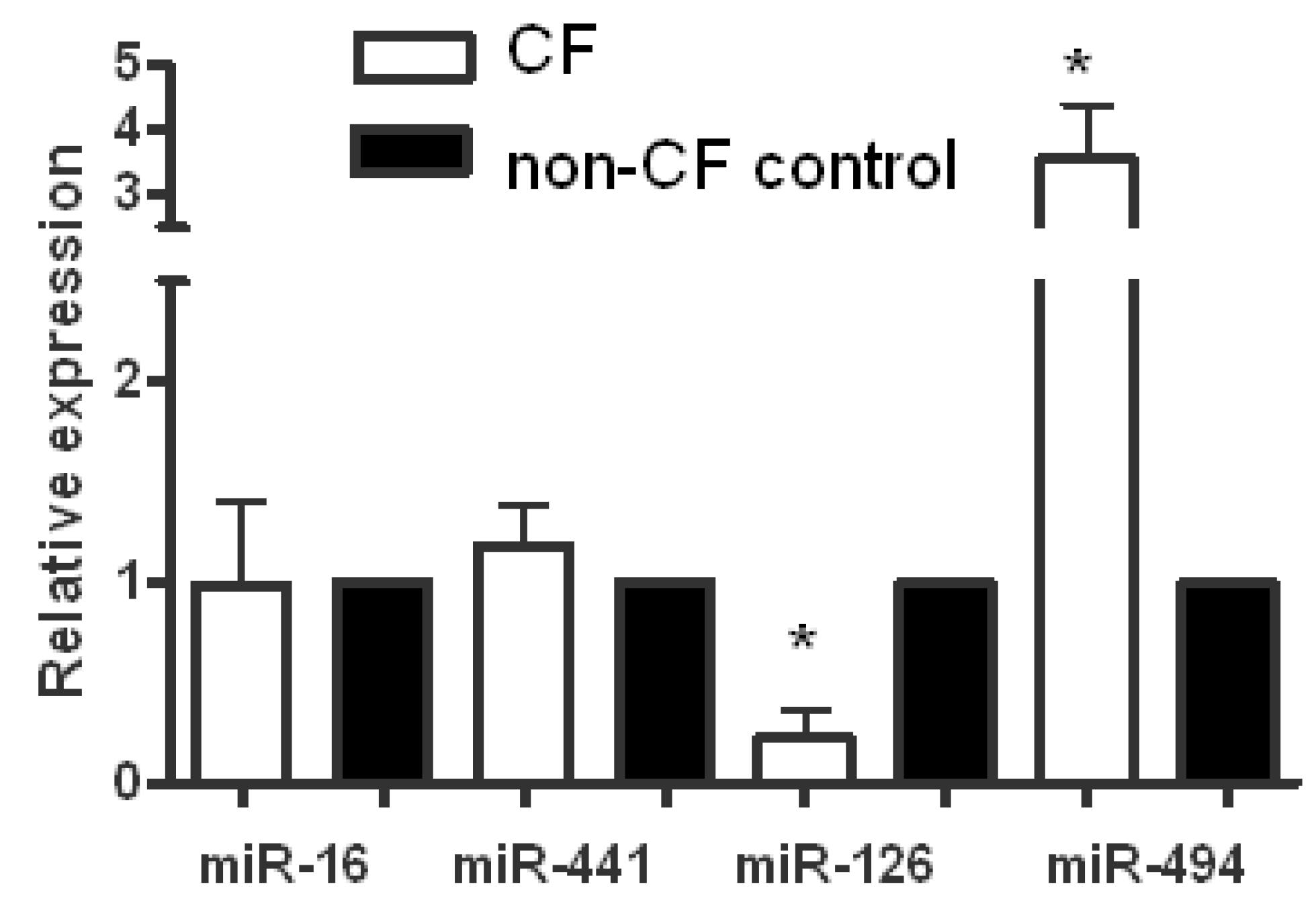
2. MicroRNA Expression Profiling Studies in Cystic Fibrosis Bronchial Epithelium

3. microRNA Regulation of CFTR Expression
4. Innate Immunity in Cystic Fibrosis
4.1. Role of miR-126 in Innate Immunity in CF Bronchial Epithelium
4.2. miR-Mediated Regulation of Interleukin-8 in CF Bronchial Epithelium
5. miR-Based Medicine for Cystic Fibrosis Lung Disease
6. Conclusion and Perspective
Acknowledgments
Conflict of Interest
References
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar]
- Lommatzsch, S.T.; Aris, R. Genetics of cystic fibrosis. Semin. Respir. Crit. Care Med. 2009, 30, 531–538. [Google Scholar] [CrossRef]
- Greene, C.M. How can we target pulmonary inflammation in cystic fibrosis? Open Respir. Med. J. 2010, 4, 18–19. [Google Scholar]
- Walsh, D.E.; Greene, C.M.; Carroll, T.P.; Taggart, C.C.; Gallagher, P.M.; O'Neill, S.J.; McElvaney, N.G. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J. Biol. Chem. 2001, 276, 35494–35499. [Google Scholar]
- Greene, C.M.; Carroll, T.P.; Smith, S.G.; Taggart, C.C.; Devaney, J.; Griffin, S.; O'Neill S, J.; McElvaney, N.G. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 2005, 174, 1638–1646. [Google Scholar]
- Bergin, D.A.; Greene, C.M.; Sterchi, E.E.; Kenna, C.; Geraghty, P.; Belaaouaj, A.; Taggart, C.C.; O'Neill, S.J.; McElvaney, N.G. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J. Biol. Chem. 2008, 283, 31736–31744. [Google Scholar]
- Chotirmall, S.H.; Greene, C.M.; Oglesby, I.K.; Thomas, W.; O'Neill, S.J.; Harvey, B.J.; McElvaney, N.G. 17beta-estradiol inhibits IL-8 in cystic fibrosis by up-regulating secretory leucoprotease inhibitor. Am. J. Respir. Crit. Care Med. 2010, 182, 62–72. [Google Scholar] [CrossRef]
- Cosgrove, S.; Chotirmall, S.H.; Greene, C.M.; McElvaney, N.G. Pulmonary proteases in the cystic fibrosis lung induce interleukin-8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/toll-like receptor pathway. J. Biol. Chem. 2011, 286, 7692–7704. [Google Scholar]
- Hassan, T.; McKiernan, P.J.; McElvaney, N.G.; Cryan, S.A.; Greene, C.M. Therapeutic modulation of miRNA for the treatment of proinflammatory lung diseases. Expert Rev. Anti Infect. Ther. 2012, 10, 359–368. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O'Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-126 is downregulated in cystic fibrosis airway epithelial cells and regulates TOM1 expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar]
- Bazett, M.; Paun, A.; Haston, C.K. MicroRNA profiling of cystic fibrosis intestinal disease in mice. Mol. Genet. Metab. 2011, 103, 38–43. [Google Scholar] [CrossRef]
- Gillen, A.E.; Gosalia, N.; Leir, S.H.; Harris, A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem. J. 2011, 438, 25–32. [Google Scholar]
- Megiorni, F.; Cialfi, S.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Synergistic post-transcriptional regulation of the cystic fibrosis transmembrane conductance regulator (CFTR) by miR-101 and miR-494 specific binding. PLoS One 2011, 6, e26601. [Google Scholar]
- Ramachandran, S.; Karp, P.H.; Jiang, P.; Ostedgaard, L.S.; Walz, A.E.; Fisher, J.T.; Keshavjee, S.; Lennox, K.A.; Jacobi, A.M.; Rose, S.D.; et al. A microRNA network regulates expression and biosynthesis of wild-type and deltaF508 mutant cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 2012, 109, 13362–13367. [Google Scholar]
- Hassan, F.; Nuovo, G.J.; Crawford, M.; Boyaka, P.N.; Kirkby, S.; Nana-Sinkam, S.P.; Cormet-Boyaka, E. miR-101 and miR-144 regulate the expression of the CFTR chloride channel in the lung. PLoS One 2012, 7, e50837. [Google Scholar]
- Oglesby, I.K.; Chotirmall, S.H.; McElvaney, N.G.; Greene, C.M. Regulation of CFTR by microRNA-145, -223 and -494 is altered in ΔF508 cystic fibrosis airway epithelium. J. Immunol. 2013. Accepted for Publication on 23rd January 2013.. [Google Scholar]
- Hartl, D.; Gaggar, A.; Bruscia, E.; Hector, A.; Marcos, V.; Jung, A.; Greene, C.; McElvaney, G.; Mall, M.; Doring, G. Innate immunity in cystic fibrosis lung disease. J. Cyst. Fibros. 2012, 11, 363–382. [Google Scholar] [CrossRef]
- Devaney, J.M.; Greene, C.M.; Taggart, C.C.; Carroll, T.P.; O'Neill, S.J.; McElvaney, N.G. Neutrophil elastase up-regulates interleukin-8 via Toll-like receptor 4. FEBS Lett. 2003, 544, 129–132. [Google Scholar] [CrossRef]
- Carroll, T.P.; Greene, C.M.; Taggart, C.C.; Bowie, A.G.; O'Neill, S.J.; McElvaney, N.G. Viral inhibition of IL-1- and neutrophil elastase-induced inflammatory responses in bronchial epithelial cells. J. Immunol. 2005, 175, 7594–7601. [Google Scholar]
- Greene, C.M.; Ramsay, H.; Wells, R.J.; O'Neill, S.J.; McElvaney, N.G. Inhibition of Toll-like receptor 2-mediated interleukin-8 production in cystic fibrosis airway epithelial cells via the alpha7-nicotinic acetylcholine receptor. Mediators Inflamm. 2010, 2010, 423241. [Google Scholar]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar]
- Keklikoglou, I.; Koerner, C.; Schmidt, C.; Zhang, J.D.; Heckmann, D.; Shavinskaya, A.; Allgayer, H.; Guckel, B.; Fehm, T.; Schneeweiss, A.; et al. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-kappaB and TGF-beta signaling pathways. Oncogene 2012, 31, 4150–4163. [Google Scholar] [CrossRef]
- Dai, L.; Gu, L.; Di, W. miR-199a attenuates endometrial stromal cell invasiveness through suppression of the IKKbeta/NF-kappaB pathway and reduced interleukin-8 expression. Mol. Hum. Reprod. 2012, 18, 136–145. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009, 1, 402–411. [Google Scholar]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Modulation of inflammatory markers by miR-146a during replicative senescence in trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 2976–2985. [Google Scholar] [CrossRef]
- Strum, J.C.; Johnson, J.H.; Ward, J.; Xie, H.; Feild, J.; Hester, A.; Alford, A.; Waters, K.M. MicroRNA-132 regulates nutritional stress-induced chemokine production through repression of sirt1. Mol. Endocrinol. 2009, 23, 1876–1884. [Google Scholar] [CrossRef]
- Hu, N.; Zhang, J.; Cui, W.; Kong, G.; Zhang, S.; Yue, L.; Bai, X.; Zhang, Z.; Zhang, W.; Zhang, X.; et al. miR-520b regulates migration of breast cancer cells by targeting hepatitis B x-interacting protein and interleukin-8. J. Biol. Chem. 2011, 286, 13714–13722. [Google Scholar]
- Yu, Z.; Willmarth, N.E.; Zhou, J.; Katiyar, S.; Wang, M.; Liu, Y.; McCue, P.A.; Quong, A.A.; Lisanti, M.P.; Pestell, R.G. MicroRNA-17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 8231–8236. [Google Scholar]
- Gambari, R.; Borgatti, M.; Bezzerri, V.; Nicolis, E.; Lampronti, I.; Dechecchi, M.C.; Mancini, I.; Tamanini, A.; Cabrini, G. Decoy oligodeoxyribonucleotides and peptide nucleic acids-DNA chimeras targeting nuclear factor kappa B: Inhibition of IL-8 gene expression in cystic fibrosis cells infected with Pseudomonas aeruginosa. Biochem. Pharmacol. 2010, 80, 1887–1894. [Google Scholar]
- Nicolis, E.; Lampronti, I.; Dechecchi, M.C.; Borgatti, M.; Tamanini, A.; Bezzerri, V.; Bianchi, N.; Mazzon, M.; Mancini, I.; Giri, M.G.; et al. Modulation of expression of IL-8 gene in bronchial epithelial cells by 5-methoxypsoralen. Int. Immunopharmacol. 2009, 9, 1411–1422. [Google Scholar]
- Cigana, C.; Nicolis, E.; Pasetto, M.; Assael, B.M.; Melotti, P. Anti-inflammatory effects of azithromycin in cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 2006, 350, 977–982. [Google Scholar] [CrossRef]
- Raia, V.; Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Auricchio, S.; Cimmino, M.; Cavaliere, M.; Nardone, M.; Cesaro, A.; et al. Inhibition of p38 mitogen activated protein kinase controls airway inflammation in cystic fibrosis. Thorax 2005, 60, 773–780. [Google Scholar] [CrossRef]
- Tchilibon, S.; Zhang, J.; Yang, Q.; Eidelman, O.; Kim, H.; Caohuy, H.; Jacobson, K.A.; Pollard, B.S.; Pollard, H.B. Amphiphilic pyridinium salts block TNF alpha/NFkappaB signaling and constitutive hypersecretion of interleukin-8 (IL-8) from cystic fibrosis lung epithelial cells. Biochem. Pharmacol. 2005, 70, 381–393. [Google Scholar]
- Fayon, M. CF-emerging therapies: Modulation inflammation. Paediatr. Respir. Rev. 2006, 8 Suppl. 1, S170–S174. [Google Scholar] [CrossRef]
- Lee, E.; Lindo, T.; Jackson, N.; Meng-Choong, L.; Reynolds, P.; Hill, A.; Haswell, M.; Jackson, S.; Kilfeather, S. Reversal of human neutrophil survival by leukotriene B(4) receptor blockade and 5-lipoxygenase and 5-lipoxygenase activating protein inhibitors. Am. J. Respir. Crit. Care Med. 1999, 160, 2079–2085. [Google Scholar]
- Esau, C.C.; Monia, B.P. Therapeutic potential for microRNAs. Adv. Drug Deliv. Rev. 2007, 59, 101–114. [Google Scholar] [CrossRef]
- Soifer, H.S.; Rossi, J.J.; Saetrom, P. MicroRNAs in disease and potential therapeutic applications. Mol. Ther. 2007, 15, 2070–2079. [Google Scholar] [CrossRef]
- Sivadas, N.; Cryan, S.A. Inhalable, bioresponsive microparticles for targeted drug delivery in the lungs. J. Pharm. Pharmacol. 2011, 63, 369–375. [Google Scholar] [CrossRef]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef]
- Clift, M.J.; Gehr, P.; Rothen-Rutishauser, B. Nanotoxicology: A perspective and discussion of whether or not in vitro testing is a valid alternative. Arch. Toxicol. 2011, 85, 723–731. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Greene, C.M. MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules 2013, 3, 157-167. https://doi.org/10.3390/biom3010157
Greene CM. MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules. 2013; 3(1):157-167. https://doi.org/10.3390/biom3010157
Chicago/Turabian StyleGreene, Catherine M. 2013. "MicroRNA Expression in Cystic Fibrosis Airway Epithelium" Biomolecules 3, no. 1: 157-167. https://doi.org/10.3390/biom3010157
APA StyleGreene, C. M. (2013). MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules, 3(1), 157-167. https://doi.org/10.3390/biom3010157