Panning for Long Noncoding RNAs

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Challenges for LncRNA Discovery
2.1. LncRNAs in General Are Expressed at Low Levels in vivo, but with High Tissue-Specificity
2.2. Evolutionary Conservation of LncRNAs on Average Is Relatively Lower than That of Coding RNAs
2.3. Controversial Coding Capacity of LncRNAs
3. Recent Progress in LncRNA Discovery Using New Strategies
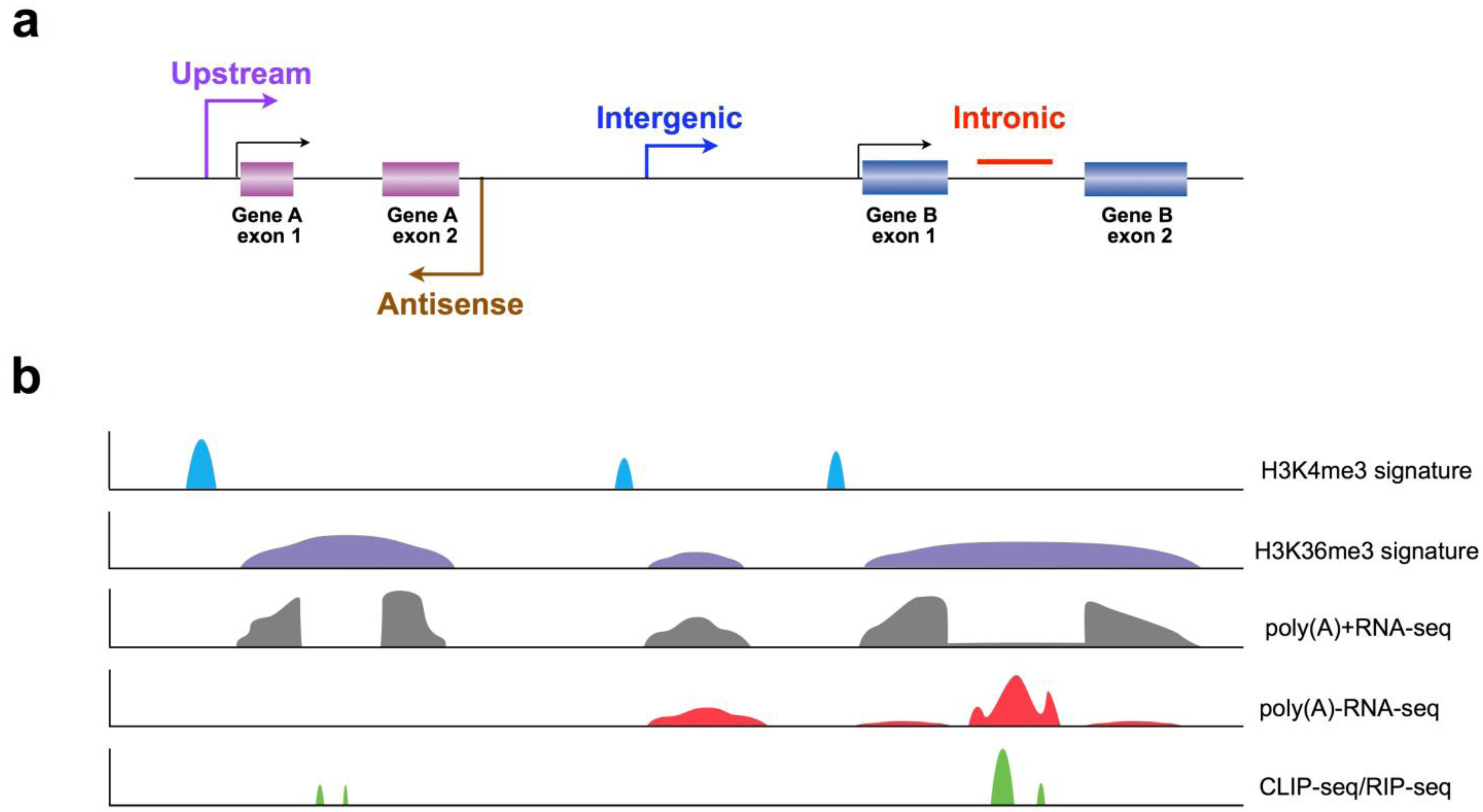
3.1. Application of Chromatin Signatures to Determine LncRNAs from Intergenic Regions
3.2. Development of a Non-Polyadenylated RNA Enrichment Strategy to Uncover LncRNAs from Introns
3.3. Determination of Co-Factors to Study LncRNA Biogenesis and Function

4. Perspectives
Acknowledgments
Conflict of Interest
References
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Yamada, K.; Lim, J.; Dale, J.M.; Chen, H.; Shinn, P.; Palm, C.J.; Southwick, A.M.; Wu, H.C.; Kim, C.; Nguyen, M.; et al. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 2003, 302, 842–846. [Google Scholar]
- Pennisi, E. Shining a light on the genome's 'dark matter'. Science 2010, 330, 1614. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; Morillon, A.; Rozowsky, J.S.; Gerstein, M.B.; Wahlestedt, C.; Hayashizaki, Y.; Carninci, P.; Gingeras, T.R.; Mattick, J.S. The reality of pervasive transcription. PLoS Biol. 2011, 9, e1000625; discussion e1001102. [Google Scholar]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Krzyzanowski, P.M.; Muro, E.M.; Andrade-Navarro, M.A. Computational approaches to discovering noncoding RNA. RNA 2012, 3, 567–579. [Google Scholar]
- Washietl, S.; Will, S.; Hendrix, D.A.; Goff, L.A.; Rinn, J.L.; Berger, B.; Kellis, M. Computational analysis of noncoding RNAs. RNA 2012, 3, 759–778. [Google Scholar]
- Chen, L.L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: what, where, and why? RNA 2010, 1, 2–21. [Google Scholar]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Mancini-Dinardo, D.; Steele, S.J.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006, 20, 1268–1282. [Google Scholar] [CrossRef]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; Chang, H.Y. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, 39. [Google Scholar]
- Chen, L.L.; Carmichael, G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell 2009, 35, 467–478. [Google Scholar] [CrossRef]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef]
- Sasaki, Y.T.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009, 19, 347–359. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; Spector, D.L. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, Franklin L.; Artandi, Steven E.; Chang, Howard Y. Genomic Maps of Long Noncoding RNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. LincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar]
- Pauli, A.; Rinn, J.L.; Schier, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136–149. [Google Scholar] [CrossRef]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.-L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12, R16. [Google Scholar] [CrossRef]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long Noncoding RNAs with snoRNA Ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar]
- Banfai, B.; Jia, H.; Khatun, J.; Wood, E.; Risk, B.; Gundling, W.E., Jr.; Kundaje, A.; Gunawardena, H.P.; Yu, Y.; Xie, L.; et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012, 22, 1646–1657. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007, 14, 103–105. [Google Scholar] [CrossRef]
- van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most "dark matter" transcripts are associated with known genes. PLoS Biol. 2010, 8, e1000371. [Google Scholar] [CrossRef]
- Johnson, J.M.; Edwards, S.; Shoemaker, D.; Schadt, E.E. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005, 21, 93–102. [Google Scholar] [CrossRef]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Mouse transcriptome: Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Hyashizaki, Y. Mouse transcriptome: Neutral evolution of ‘non-coding’ complementary DNAs (reply). Nature 2004, 431. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Zheng, H.; Li, J.; Liu, D.; Li, H.; Samudrala, R.; Yu, J.; Wong, G.K. Neutral evolution of ‘non-coding’ complementary DNAs. Nature 2004, 431. [Google Scholar] [CrossRef]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef]
- Marques, A.C.; Tan, J.; Lee, S.; Kong, L.; Heger, A.; Ponting, C.P. Evidence for conserved post-transcriptional roles of unitary pseudogenes and for frequent bifunctionality of mRNAs. Genome Biol. 2012, 13, R102. [Google Scholar] [CrossRef]
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: an answer to the 'genome complexity' conundrum. Genes Dev. 2007, 21, 11–42. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef]
- Imanishi, T.; Itoh, T.; Suzuki, Y.; O'Donovan, C.; Fukuchi, S.; Koyanagi, K.O.; Barrero, R.A.; Tamura, T.; Yamaguchi-Kabata, Y.; Tanino, M.; et al. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol. 2004, 2, e162. [Google Scholar]
- Duret, L.; Chureau, C.; Samain, S.; Weissenbach, J.; Avner, P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 2006, 312, 1653–1655. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef]
- Chen, L.L.; Carmichael, G.G. Decoding the function of nuclear long non-coding RNAs. Curr. Opin. Cell Biol. 2010, 22, 357–364. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef]
- Kloc, M.; Wilk, K.; Vargas, D.; Shirato, Y.; Bilinski, S.; Etkin, L.D. Potential structural role of non-coding and coding RNAs in the organization of the cytoskeleton at the vegetal cortex of Xenopus oocytes. Development 2005, 132, 3445–3457. [Google Scholar]
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3' end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef]
- Cui, P.; Lin, Q.; Ding, F.; Xin, C.; Gong, W.; Zhang, L.; Geng, J.; Zhang, B.; Yu, X.; Yang, J.; Hu, S.; Yu, J. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010, 96, 259–265. [Google Scholar]
- Gardner, E.J.; Nizami, Z.F.; Talbot, C.C., Jr.; Gall, J.G. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 2012, 26, 2550–2559. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Jnbaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3' ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3'-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar]
- Box, J.A.; Bunch, J.T.; Tang, W.; Baumann, P. Spliceosomal cleavage generates the 3' end of telomerase RNA. Nature 2008, 456, 910–914. [Google Scholar] [CrossRef]
- Seitz, H. Redefining microRNA targets. Curr. Biol. 2009, 19, 870–873. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Feng, J.; Bi, C.; Clark, B.S.; Mady, R.; Shah, P.; Kohtz, J.D. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006, 20, 1470–1484. [Google Scholar] [CrossRef]
- Schmitt, S.; Prestel, M.; Paro, R. Intergenic transcription through a polycomb group response element counteracts silencing. Genes Dev. 2005, 19, 697–708. [Google Scholar] [CrossRef]
- Sanchez-Elsner, T.; Gou, D.; Kremmer, E.; Sauer, F. Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science 2006, 311, 1118–1123. [Google Scholar] [CrossRef]
- Zappulla, D.C.; Cech, T.R. Yeast telomerase RNA: a flexible scaffold for protein subunits. Proc. Natl. Acad. Sci. USA 2004, 101, 10024–10029. [Google Scholar] [CrossRef]
- Mao, Y.S.; Sunwoo, H.; Zhang, B.; Spector, D.L. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 2011, 13, 95–101. [Google Scholar] [CrossRef]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal 2010, 3, ra8. [Google Scholar] [CrossRef]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar]
- Hasegawa, Y.; Brockdorff, N.; Kawano, S.; Tsutui, K.; Nakagawa, S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 2010, 19, 469–476. [Google Scholar] [CrossRef]
- Yeo, G.W.; Coufal, N.G.; Liang, T.Y.; Peng, G.E.; Fu, X.-D.; Gage, F.H. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009, 16, 130–137. [Google Scholar] [CrossRef]
- Wan, Y.; Qu, K.; Ouyang, Z.; Kertesz, M.; Li, J.; Tibshirani, R.; Makino, D.L.; Nutter, R.C.; Segal, E.; Chang, H.Y. Genome-wide Measurement of RNA Folding Energies. Mol. Cell 2012, 48, 169–181. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, S.; Zhang, X.-O.; Yang, L. Panning for Long Noncoding RNAs. Biomolecules 2013, 3, 226-241. https://doi.org/10.3390/biom3010226
Zhu S, Zhang X-O, Yang L. Panning for Long Noncoding RNAs. Biomolecules. 2013; 3(1):226-241. https://doi.org/10.3390/biom3010226
Chicago/Turabian StyleZhu, Shanshan, Xiao-Ou Zhang, and Li Yang. 2013. "Panning for Long Noncoding RNAs" Biomolecules 3, no. 1: 226-241. https://doi.org/10.3390/biom3010226
APA StyleZhu, S., Zhang, X. -O., & Yang, L. (2013). Panning for Long Noncoding RNAs. Biomolecules, 3(1), 226-241. https://doi.org/10.3390/biom3010226