
Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease

{kind=link}
{kind=link}
Abstract
:1. Pathological Characteristics and Aetiology of Parkinson’s Disease
2. Parkinson’s Disease and α-Synuclein
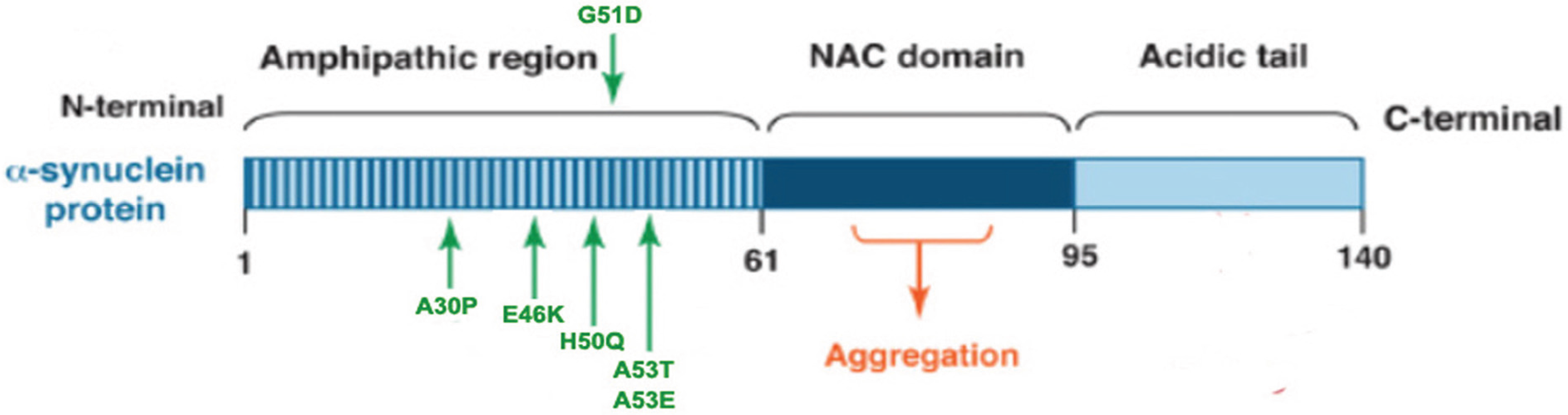
2.1. α-Synuclein is Linked to the Pathogenesis of Parkinson’s Disease
2.2. α-Synuclein and Cytotoxicity

2.3. α-Synuclein Post-Translational Modifications
2.4. α-Synuclein and Oxidative Stress
3. Parkinson’s Disease and Neuromelanin
3.1. Neuromelanin Structure and Biosynthesis
3.2. Neuromelanin Involved in the Pathogenesis of Parkinson’s Disease
3.3. Interaction of Neuromelanin with Organic or Inorganic Molecules
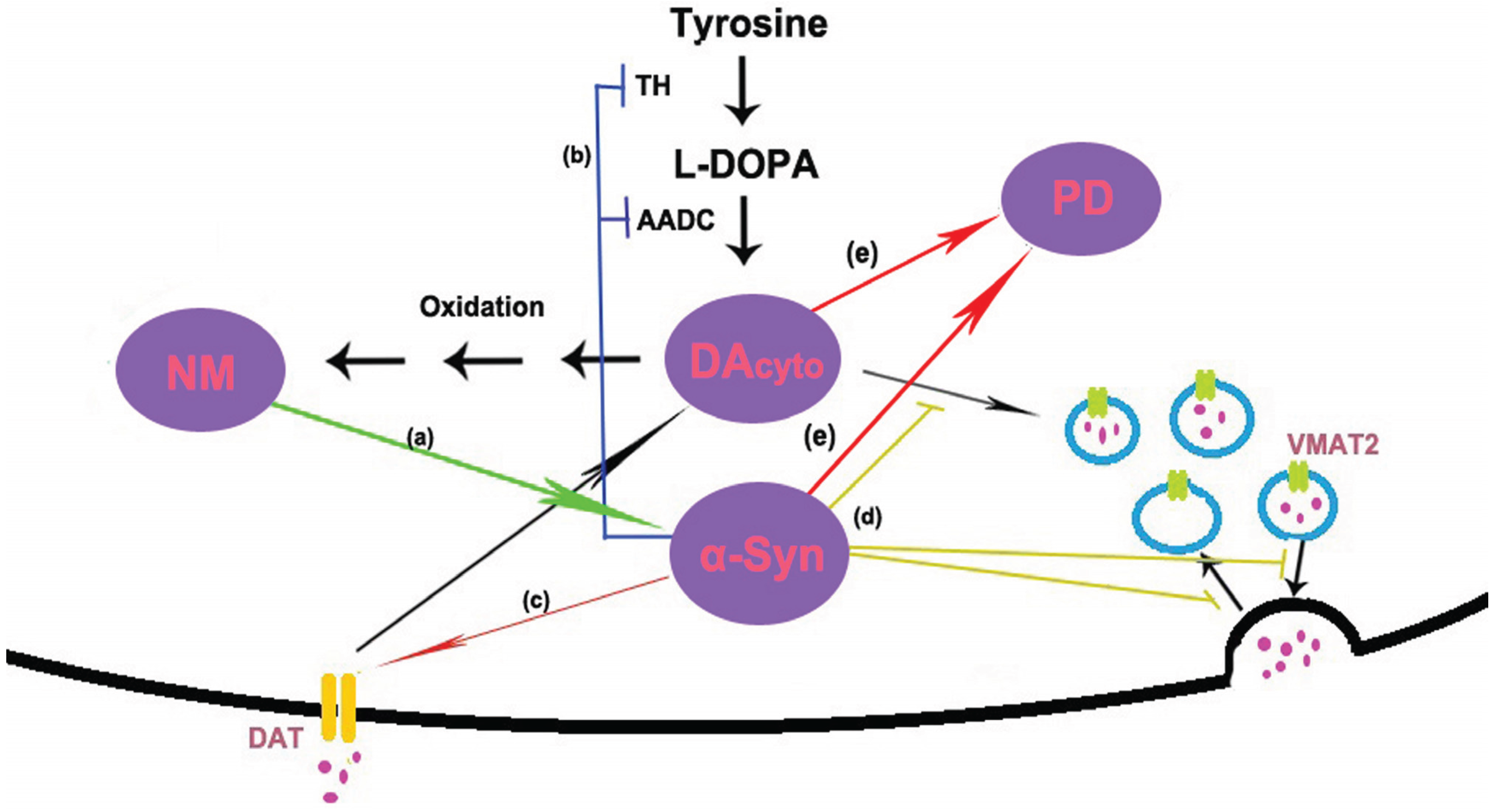
4. Interaction between α-Synuclein and Neuromelanin
4.1. Accumulation of Nenromelanin Increases the Level of α-Synuclein in SN Neurons
4.2. α-Synuclein Induces the Biosynthesis of Neuromelanin
5. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 2004, 1, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 7, 229–238. [Google Scholar]
- Gonera, E.G.; Van’t Hof, M.; Berger, H.J.; van Weel, C.; Horstink, M.W. Symptoms and duration of the prodromal phase in Parkinson’s disease. Mov. Disord. 1997, 12, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Reichmann, H. Non-motor dysfunction in Parkinson’s disease. Park. Relat. Disord. 2007, 13, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Maass, A.; Reichmann, H. Sleep and non-motor symptoms in Parkinson’s disease. J. Neural Transm. 2013, 120, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Morris, H.R.; Robbins, T.W.; Goedert, M.; Hardy, J.; Ben-Shlomo, Y.; Bolam, P.; Burn, D.; Hindle, J.V.; Brooks, D. Parkinson’s disease—The debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J. Parkinsons Dis. 2013, 3, 1–11. [Google Scholar] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Westbroek, W.; Gustafson, A.M.; Sidransky, E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 2011, 17, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Gouider-Khouja, N.; Larnaout, A.; Amouri, R.; Sfar, S.; Belal, S.; Ben Hamida, C.; Ben Hamida, M.; Hattori, N.; Mizuno, Y.; Hentati, F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Park. Relat. Disord. 2003, 9, 247–251. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Skipper, L.; Wilkes, K.; Toft, M.; Baker, M.; Lincoln, S.; Hulihan, M.; Ross, O.A.; Hutton, M.; Aasly, J.; Farrer, M. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am. J. Hum. Genet. 2004, 75, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Wavrant-De Vrieze, F.; Crook, R.; Boles, L.; Perez-Tur, J.; Hardy, J.; Johnson, W.G.; Steele, J.; Maraganore, D.; Gwinn, K.; et al. Low frequency of alpha-synuclein mutations in familial Parkinson’s disease. Ann. Neurol. 1998, 43, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein cause selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Park. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal pigmented autophagic vacuoles: Lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J. Neurochem. 2008, 106, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Q.; Xu, S.L.; Lu, D.H.; Yu, S.; Zhou, M.; Uéda, K.; Cui, Y.Q.; Zhang, B.Y.; Chan, P. Increased expression of α-synuclein in aged human brain associated with neuromelanin accumulation. J. Neural Transm. 2011, 118, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Bishop, J.F.; Eglitis, M.A.; Yang, Y.; Mouradian, M.M. Enhanced viability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience 2000, 97, 279–284. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances α-synuclein aggregation, secretion and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 18, 145–175. [Google Scholar]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.Y.; Tang, Z.; Liu, C.W. α-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: Understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J. Biol. Chem. 2008, 283, 20288–20298. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Grillo-Bosch, D. In vitro preparation of prefibrillar intermediates of amyloid-beta and alpha-synuclein. Methods Mol. Biol. 2005, 299, 19–33. [Google Scholar] [PubMed]
- Cole, N.B.; Murphy, D.D.; Lebowitz, J.; Di Noto, L.; Levine, R.L.; Nussbaum, R.L. Metal-catalyzed oxidation of alpha-synuclein: Helping to define the relationship between oligomers, protofibrils, and filaments. J. Biol. Chem. 2005, 280, 9678–9690. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Fagerqvist, T.; Lindström, V.; Nordström, E.; Lord, A.; Tucker, S.M.; Su, X.; Sahlin, C.; Kasrayan, A.; Andersson, J.; Welander, H.; et al. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J. Neurochem. 2013, 126, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, S.J. Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J. Biol. Chem. 2002, 277, 48976–48983. [Google Scholar] [CrossRef] [PubMed]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lavedan, C. The synuclein family. Genome Res. 1998, 8, 871–880. [Google Scholar] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- George, J.M. The synucleins. Genome Biol. 2002. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T. Accelerated oligomerization by Parkinson’s disease linked α-synuclein mutants. Ann. NY Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the α-synuclein protofibril by a dopamine—α-Synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [PubMed]
- Jethva, P.N.; Kardani, J.R.; Roy, I. Modulation of α-synuclein aggregation by dopamine in the presence of MPTP and its metabolite. FEBS J. 2011, 278, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Jenco, J.M.; Rawlingson, A.; Daniels, B.; Morris, A.J. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell. 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar]
- Oueslati, A.; Ximerakis, M.; Vekrellis, K. Protein transmission, seeding and degradation: key steps for α-synuclein prion-like propagation. Exp. Neurobiol. 2014, 23, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New α- and γ-synuclein immunopathological lesions in human brain. Acta Neuropathol Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [PubMed]
- Takahashi, T.; Yamashita, H.; Nagano, Y.; Nakamura, T.; Ohmori, H.; Avraham, H.; Avraham, S.; Yasuda, M.; Matsumoto, M. Identification and characterization of a novel Pyk2/related adhesion focal tyrosine kinase-associated protein that inhibits alpha-synuclein phosphorylation. J. Biol. Chem. 2003, 278, 42225–42233. [Google Scholar] [CrossRef] [PubMed]
- Azeredo da Silveira, S.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of α-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887. [Google Scholar] [PubMed]
- McFarland, N.R.; Fan, Z.; Xu, K.; Schwarzschild, M.A.; Feany, M.B.; Hyman, B.T.; McLean, P.J. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2009, 68, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: Areview of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 2008, 67, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Bisharat, S.; Engelender, S. α-Synuclein ubiquitination and novel therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yahya, M.; Fauvet, B.; Herman-Bachinsky, Y.; Hejjaoui, M.; Bavikar, S.N.; Karthikeyan, S.V.; Ciechanover, A.; Lashuel, H.A.; Brik, A. Synthetic polyubiquitinated α-synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, 17726–17731. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Garcia Reitböck, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein (1–120): Implications for Lewy body disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. α-Synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Hashimoto, M.; Takeda, A.; Hsu, L.J.; Takenouchi, T.; Masliah, E. Role of cytochrome C as a stimulator of alpha-synuclein aggregation in Lewy body disease. J. Biol. Chem. 1999, 274, 28849–28852. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Högen, T.; Hillmer, A.S.; Bader, B.; Schmidt, F.; Kamp, F.; Kretzschmar, H.A.; Bötzel, K.; Giese, A. Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Park. Dis. 2011, 1, 205–216. [Google Scholar] [CrossRef]
- Kim, K.S.; Choi, S.Y.; Kwon, H.Y.; Won, M.H.; Kang, T.C.; Kang, J.H. Aggregation of α-synuclein induced by the Cu, Zn-superoxide dismutase and hydrogen peroxide system. Free Radic. Biol. Med. 2002, 32, 544–550. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, M.; Yu, S.; Cai, Y.; Zhang, A.; Uéda, K.; Chan, P. Oxidative stress induces nuclear translocation of C-terminus of alpha-synuclein in dopaminergic cells. Biochem. Biophys. Res. Commun. 2006, 342, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, S.; Mi, J.; Uéda, K.; Chan, P. Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res. 2013, 1500, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Matsuzaki-Kobayashi, M.; Takeda, A.; Sugeno, N.; Kikuchi, A.; Furukawa, K.; Perry, G.; Smith, M.A.; Itoyama, Y. Alpha-synuclein facilitates the toxicity of oxidized catechol metabolites: Implications for selective neurodegeneration in Parkinson’s disease. FEBS Lett. 2006, 580, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Uéda, K.; Chan, P. α-Synuclein and dopamine metabolism. Mol. Neurobiol. 2005, 31, 243–254. [Google Scholar] [CrossRef]
- Schlüter, O.M.; Fornai, F.; Alessandrí, M.G.; Takamori, S.; Geppert, M.; Jahn, R.; Südhof, T.C. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 2003, 118, 985–1002. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, S.L.; Chen, B. Dual effects of different concentrations of alpha-synuclein on the neurotoxicity of 6-hydroxydopamine in SH-SY5Y cells. Sheng Li Xue Bao 2009, 61, 324–330. [Google Scholar] [PubMed]
- Thomas, B.; Mandir, A.S.; West, N.; Liu, Y.; Andrabi, S.A.; Stirling, W.; Dawson, V.L.; Dawson, T.M.; Lee, M.K. Resistance to MPTP-neurotoxicity in α-synuclein knockout mice is complemented by human α-synuclein and associated with increased β-synuclein and Akt activation. PLoS ONE 2011, 6, e16706. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.E.; Tennyson, V.M. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus coeruleus in Parkinson’s disease. J. Neuropthol. Exp. Neurol. 1965, 24, 398–414. [Google Scholar] [CrossRef]
- Fenichel, G.M.; Bazelon, M. Studies on neuromelanin. II. Melanin in the brainstems of infants and children. Neurology 1968, 18, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Moss, S.C.; Eisner, M. X-ray characterization of melanins–II. Pigment Cell Res. 1994, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Haavik, J. L-DOPA is a substrate for tyrosine hydroxylase. J. Neurochem. 1997, 69, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.R. The role of peroxidase in neuromelanin synthesis: A review. Physiol. Chem. Phys. Med. NMR 1997, 29, 15–22. [Google Scholar] [PubMed]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, R.J.; Lipman, Z.P.; Snyder, S.H. Selectivity of the parkinsonian neurotoxin MPTP: Toxic metabolite MPP+ binds to neuromelanin. Science 1986, 231, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, N.G.; Larsson, B.S.; Lydén-Sokolowski, A. Neuromelanin and its possible protective and destructive properties. Pigment Cell Res. 1987, 1, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Pietra, R.; Goj, C.; Mecacci, C.; Radice, D.; Sabbioni, E. Iron and other metals in neuromelanin, substantia nigra, and putamen of human brain. J. Neurochem. 1994, 62, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Fornstedt, B.; Brun, A.; Rosengren, E.; Carlsson, A. The apparent autoxidation rate of catechols in dopamine-rich regions of human brains increases with the degree of depigmentation of substantia nigra. J. Neural Transm. Park. Dis. Dement Sect. 1989, 1, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Wilczok, T.; Stepien, K.; Dzierzega-Lecznar, A.; Zajdel, A.; Wilczok, A. Model neuromelanins as antioxidative agents during lipid peroxidation. Neurotox. Res. 1999, 1, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Shamoto-Nagai, M.; Maruyama, W.; Akao, Y.; Osawa, T.; Tribl, F.; Gerlach, M.; Zucca, F.A.; Zecca, L.; Riederer, P.; Naoi, M. Neuromelanin inhibits enzymatic activity of 26S proteasome in human dopaminergic SH-SY5Y cells. J. Neural Transm. 2004, 111, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Zareba, M.; Bober, A.; Korytowski, W.; Zecca, L.; Sarna, T. The effect of a synthetic neruomelanin on yield of free hydroxyl radicals generated in model systems. Biochim. Biophys. Acta 1995, 1271, 343–348. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemical degradation of melanins: Application to identification of dopamine-melanin. Pigment Cell Res. 1998, 11, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Ophof, A.; Broe, M.; Jensen, P.H.; Kettle, E.; Fedorow, H.; Cartwright, M.I.; Griffiths, F.M.; Shepherd, C.E.; Double, K.L. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 2005, 128, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, Z.F.; Gao, J.H.; Sun, L.; Huang, X.Y.; Liu, Z.; Yu, S.Y.; Cao, C.J.; Zuo, L.J.; Chen, Z.J.; et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: Implications for Parkinson’s disease. FASEB J. 2003, 17, 500–502. [Google Scholar] [PubMed]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ostergren, A.; Annas, A.; Skog, K.; Lindquist, N.G.; Brittebo, E.B. Longterm retention of neurotoxic beta-carbolines in brain neuromelanin. J. Neural Transm. 2004, 111, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, N.G.; Larsson, B.S.; Lyden-Sokolowski, A. Autoradiography of [14C]paraquat or [14C]diquat in frogs and mice: Accumulation in neuromelanin. Neurosci. Lett. 1988, 93, 1–6. [Google Scholar] [CrossRef]
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulzer, D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pigment Cell Res. 2004, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Mecacci, C.; Seraglia, R.; Parati, E. The chemical characterization of melanin contained in substantia nigra of human brain. Biochim. Biophys. Acta 1992, 1138, 6–10. [Google Scholar] [CrossRef]
- Zecca, L.; Costi, P.; Mecacci, C.; Ito, S.; Terreni, M.; Sonnino, S. Interaction of human substantia nigra neuromelanin with lipids and peptides. J. Neurochem. 2000, 74, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Fasano, M.; Bergamasco, B.; Lopiano, L.; Valente, G. Evidence for a glycidic-lipidic matrix in human neuromelanin, potentially responsible for the enhanced iron sequestering ability of substantia nigra. J. Neurochem. 1994, 62, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Barden, H. The histochemical relationship of neuromelanin and lipofuscin. J. Neuropathol. Exp. Neurol. 1969, 28, 419–441. [Google Scholar] [CrossRef] [PubMed]
- Swartz, H.M.; Sarna, T.; Zecca, L. Modulation by neuromelanin of the availability and reactivity of metal ions. Ann. Neurol. 1992, 32, S69–S75. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Bellei, C.; Giannelli, S.; Terreni, M.R.; Gallorini, M.; Rizzio, E.; Pezzoli, G.; Albertini, A.; Zecca, L. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: Consequences for neuronal vulnerability. J. Neural. Transm. 2006, 113, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2003, 86, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Gerlach, M.; Schunemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.; Riederer, P.; Ben-Shachar, D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for Parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Gründemann, J.; Schlaudraff, F.; Liss, B. UV-laser microdissection and mRNA expression analysis of individual neurons from postmortem Parkinson’s disease brains. Methods Mol. Biol. 2011, 755, 363–374. [Google Scholar] [PubMed]
- Matsuo, Y.; Kamitani, T. Parkinson’s disease-related protein, α-synuclein, in malignant melanoma. PLoS ONE 2010, 5, e10481. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Giraudo, S.; Coha, S.; Bergamasco, B.; Lopiano, L. Residual substantia nigra neuromelanin in Parkinson’s disease is cross-linked to alpha-synuclein. Neurochem. Int. 2003, 42, 603–606. [Google Scholar] [CrossRef]
- Fedorow, H.; Halliday, G.M.; Rickert, C.H.; Gerlach, M.; Riederer, P.; Double, K.L. Evidence for specific phases in the development of human neuromelanin. Neurobiol. Aging 2006, 27, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Ben-Shachar, D.; Youdim, M.B.; Zecca, L.; Riederer, P.; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol. 2002, 24, 621–628. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Yi, H.; Akao, Y.; Tribl, F.; Gerlach, M.; Osawa, T.; Riederer, P.; Naoi, M. Neuromelanin induces oxidative stress in mitochondria through release of iron: Mechanism behind the inhibition of 26S proteasome. J. Neural Transm. 2006, 113, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Zhu, J.; Hwu, W.J.; Jankovic, J. The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS ONE 2012, 7, e45183. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Zucca, F.A.; Costi, P.; Tampellini, D.; Gatti, A.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Ito, S.; Gallorini, M.; et al. The neuromelanin of human substantia nigra: Structure, synthesis and molecular behaviour. J. Neural Transm. Suppl. 2003, 65, 145–155. [Google Scholar] [PubMed]
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Tessari, I.; Bisaglia, M.; Valle, F.; Samorì, B.; Bergantino, E.; Mammi, S.; Bubacco, L. The reaction of alpha-synuclein with tyrosinase: Possible implications for Parkinson disease. J. Biol. Chem. 2008, 283, 16808–16817. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Gao, N.; Ye, Y.W.; Li, X.; Yu, S.; Yang, H.; Uéda, K.; Chan, P. α-Synuclein functions as a negative regulator for expression of tyrosine hudroxylase. Acta Neurol. Belg. 2011, 111, 130–135. [Google Scholar] [PubMed]
- Koo, H.J.; Yang, J.E.; Park, J.H.; Lee, D.; Paik, S.R. α-Synuclein-mediated defense against oxidative stress via modulation of glutathione peroxidase. Biochim. Biophys. Acta 2013, 1834, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Fornstedt, B.; Rosengren, E.; Carlsson, A. Occurrence and distribution of 5-S-cysteinyl derivatives of dopamine, dopa and dopac in the brains of eight mammalian species. Neuropharmacology 1986, 25, 451–454. [Google Scholar] [CrossRef]
- Liang, C.L.; Nelson, O.; Yazdani, U.; Pasbakhsh, P.; German, D.C. Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: Human midbrain dopamine neurons. J. Comp. Neurol. 2004, 473, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zuo, X.; Li, Y.; Zhang, C.; Zhou, M.; Zhang, Y.A.; Uéda, K.; Chan, P. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 2004, 367, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Falarti, E.; Zaltieri, M.; Bono, F.; Collo, G.; Spillantini, M.G.; Missale, C.; Spano, P. Redistribution of DAT/α-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS ONE 2011, 6, e27959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oaks, A.W.; Marsh-Armstrong, N.; Jones, J.M.; Credle, J.J.; Sidhu, A. Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS ONE 2013, 8, e70872. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Staal, R.G.; Bove, J.; Prou, D.; Hananiya, A.; Markov, D.; Poulsen, N.; Larsen, K.E.; Moore, C.M.; Troyer, M.D.; et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J. Neurosci. 2006, 26, 9304–9311. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Chow, A.M.; Cheng, X.R.; Tang, D.W.; Brown, I.R.; Kerman, K. Oxidative stress effect of dopamine on α-synuclein: Electroanalysis of solvent interactions. ACS Chem. Neurosci. 2012, 3, 569–574. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Chan, P. Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease. Biomolecules 2015, 5, 1122-1142. https://doi.org/10.3390/biom5021122
Xu S, Chan P. Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease. Biomolecules. 2015; 5(2):1122-1142. https://doi.org/10.3390/biom5021122
Chicago/Turabian StyleXu, Shengli, and Piu Chan. 2015. "Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease" Biomolecules 5, no. 2: 1122-1142. https://doi.org/10.3390/biom5021122
APA StyleXu, S., & Chan, P. (2015). Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease. Biomolecules, 5(2), 1122-1142. https://doi.org/10.3390/biom5021122