
The Altered Hepatic Tubulin Code in Alcoholic Liver Disease

Abstract
:1. Introduction
2. The Tubulin Code
2.1. Modifications of the Tubulin Code
2.2. Acetylation Is the Primary Modification in Hepatocytes and It Is Enhanced upon Ethanol Exposure

{kind=link}
{kind=link}
{kind=link}
| Major PTM | α/β | Site | Distribution | References | Hepatocytes | References |
|---|---|---|---|---|---|---|
| Acetylation | α | Lys40 | Centrioles, midbodies, mitotic spindles, neurons, cilia, flagella, cytoplasmic microtubules | [27,28,29,30,31,32,33] | Cytoplasmic microtubules | [17,20] |
| β | Lys252 | Soluble dimer | [34] | |||
| Detyrosination | α | C-terminal Tyr removal | Centrioles, midbodies, mitotic spindles, neurons, cilia, flagella, cytoplasmic microtubules | [28,29,35,36,37,38,39,40,41,42] | Centrioles (?) | [17] |
| Deglutamylation (Δ2-tubulin) | α | C-terminal Glu removal from detryrosinated CTTs | Centrioles, neurons, cilia, flagella | [43,44,45,46] | ||
| Mono/poly-Glutamylation | α/β | Glu(s) addition to Glu in CTTs | Centrioles, midbodies, mitotic spindles, neurons, cilia, flagella, cytoplasmic microtubules (mono only) | [47,48,49,50,51,52,53,54,55,56,57] | Centrioles (?) | [17] |
| Mono/poly-Glycylation | α/β | Gly(s) addition to Glu in CTTs | Cilia, flagella | [35,58,59,60] | ||
| Minor PTM | Comments | References | ||||
| Polyamination | Found only in neurons; Gln15 in β-tubulin and other unidentified α- and β-tubulin sites | [15] | ||||
| O-linked Glycosylation | Examined only in neurons, B lymphocytes, HeLa cells, L6 myotubes and MN9D neuronal cells; various unidentified α- and β-tubulin sites | [61,62,63,64] | ||||
| Palmitoylation | Examined only in neurons (Cys376 in α-tubulin) and in yeast (Cys377 in α-tubulin) | [65,66,67] | ||||
| Phosphorylation | Examined only in neuroblastoma cells, rat brain and COS-7 cells; various unidentified α- and β-tubulin sites and Ser172 in soluble β-tubulin | [68,69,70,71,72] | ||||
| Sumoylation | Examined only in yeast and HEK293 cells (overexpressing SUMO); multiple unidentified α-tubulin Lys | [73,74] | ||||
| Ubiquitination | Examined only in neurons, cilia, flagella, and HEK293 cells (overexpressing Parkin); multiple unidentified α-tubulin Lys | [75,76,77] | ||||
| Succination | Examined in adipocytes, C2C12 myotubes grown in high glucose and adipose tissue of db/db diabetic mice; Cys347 and 376 in α-tubulin, Cys12 and 303 in β-tubulin | [78] | ||||
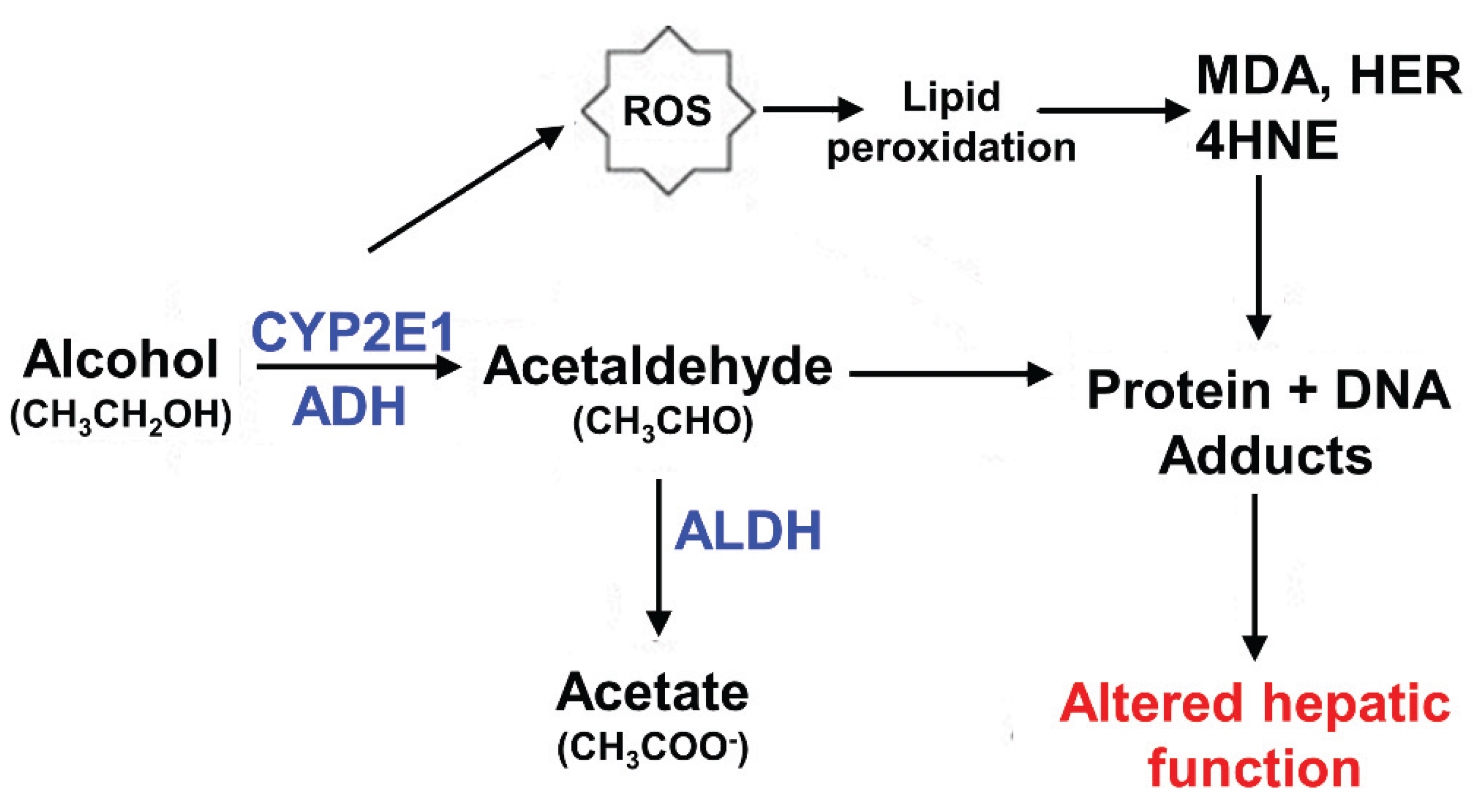
2.3. Other Ethanol-Induced Modifications of Microtubules

2.4. A Possible Mechanisms for Ethanol-Induced Microtubule Acetylation
3. Consequences of Altered Microtubule Modifications on Cellular Function
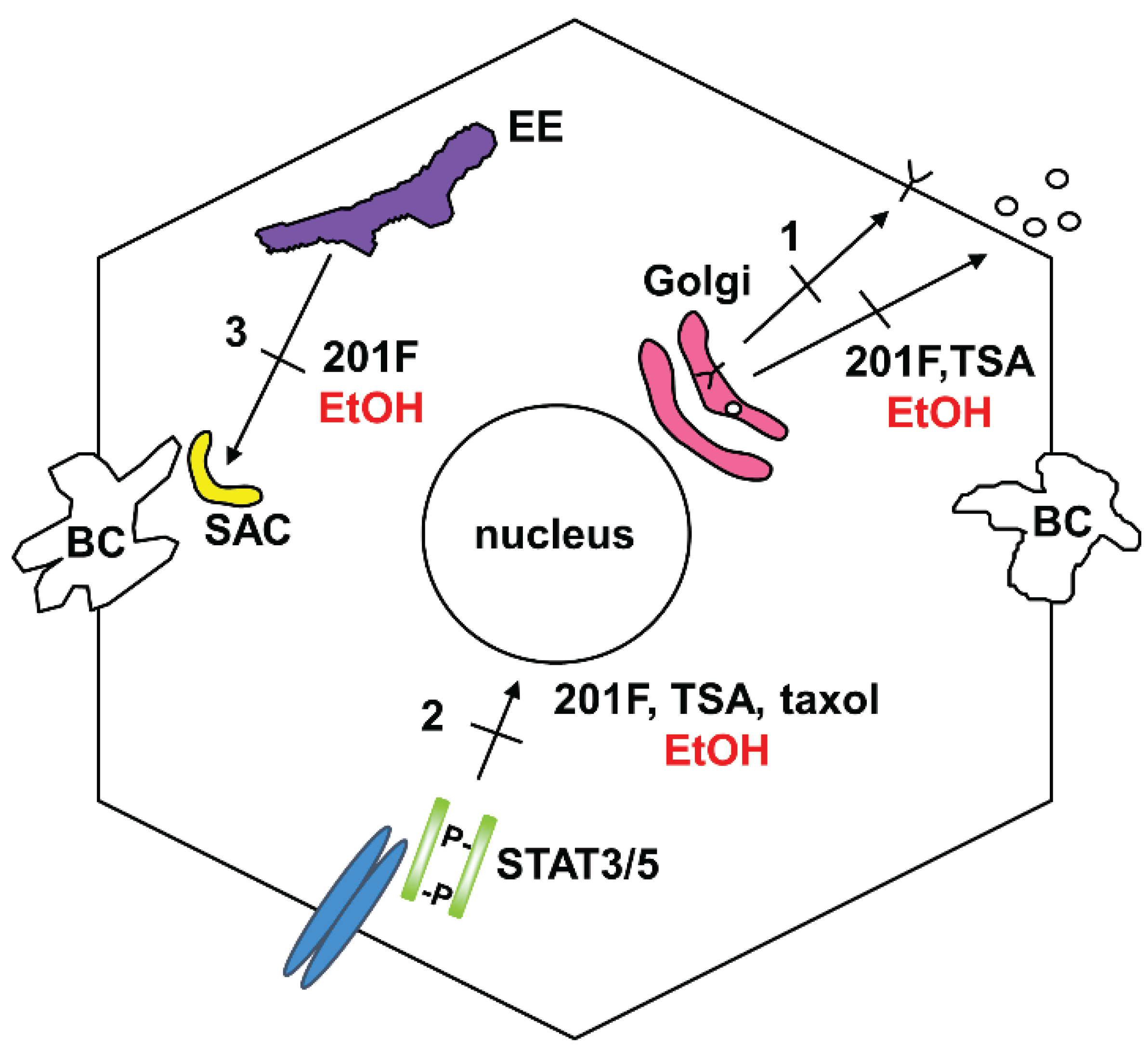
3.1. Impaired Protein Trafficking

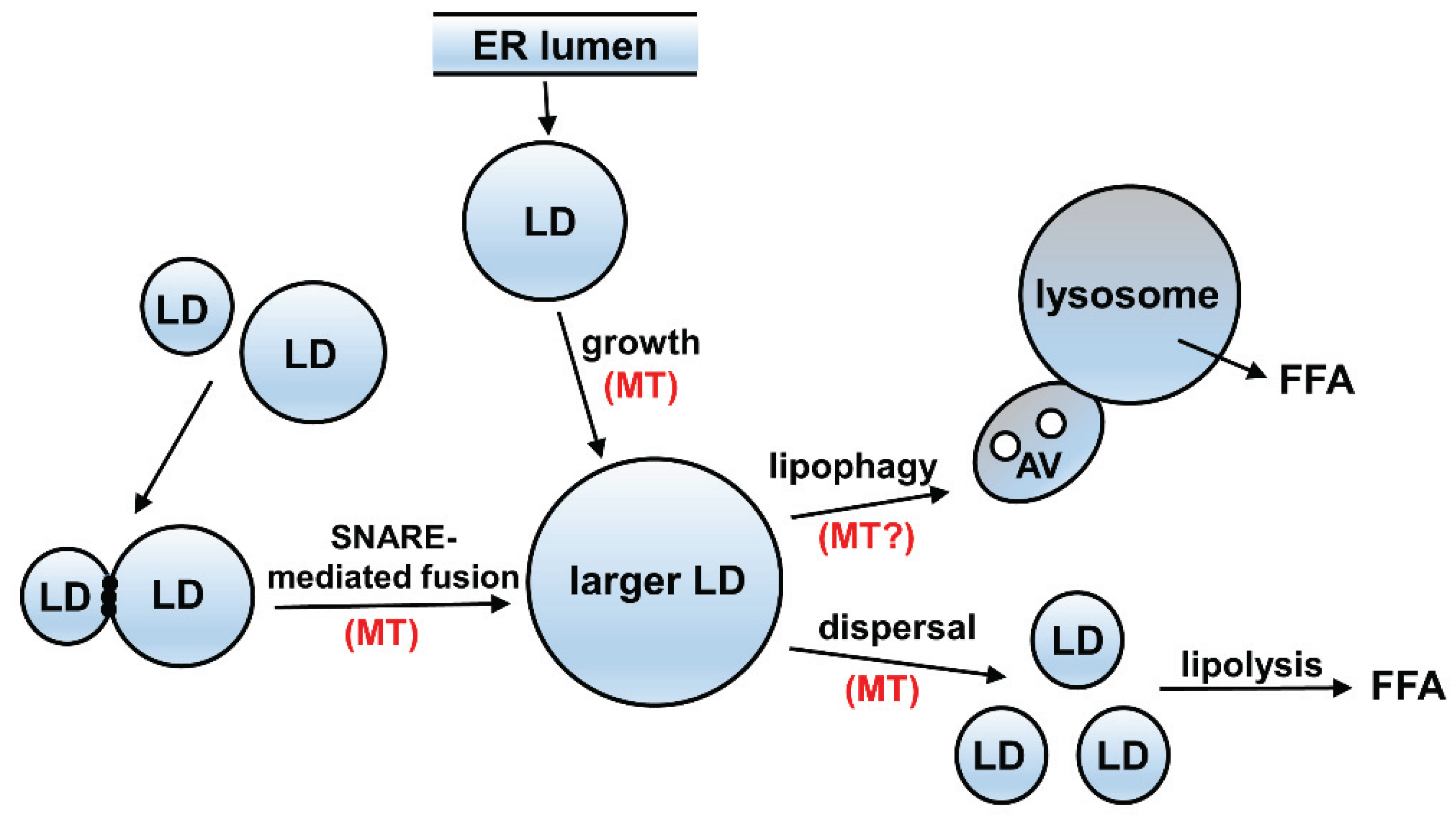
3.2. A Possible Relationship between Acetylated Microtubules and Alcohol-Induced Steatosis

4. Possible Mechanism of Impaired Microtubule-Mediated Processes
5. Clinical Significance of Altered Microtubule Post-Modifications and Potential Therapeutics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Status Report on Alcohol and Health; World Health Organization: Geneva, Switzerland, 2014; pp. 48–51. [Google Scholar]
- Jennett, R.B.; Sorrell, M.F.; Johnson, E.L.; Tuma, D.J. Covalent binding of acetaldehyde to tubulin: Evidence for preferential binding to the alpha-chain. Arch. Biochem. Biophys. 1987, 256, 10–18. [Google Scholar] [CrossRef]
- Jennett, R.B.; Sorrell, M.F.; Saffari-Fard, A.; Ockner, J.L.; Tuma, D.J. Preferential covalent binding of acetaldehyde to the alpha-chain of purified rat liver tubulin. Hepatology 1989, 9, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Szasz, J.; Burns, R.; Sternlicht, H. Effects of reductive methylation on microtubule assembly. Evidence for an essential amino group in the alpha-chain. J. Biol. Chem. 1982, 257, 3697–3704. [Google Scholar] [PubMed]
- Szasz, J.; Yaffe, M.B.; Elzinga, M.; Blank, G.S.; Sternlicht, H. Microtubule assembly is dependent on a cluster of basic residues in alpha-tubulin. Biochemistry 1986, 25, 4572–4582. [Google Scholar] [CrossRef] [PubMed]
- Tuma, D.J.; Jennett, R.B.; Sorrell, M.F. The interaction of acetaldehyde with tubulin. Ann. NY Acad. Sci. 1987, 492, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Tuma, D.J.; Smith, S.L.; Sorrell, M.F. Acetaldehyde and microtubules. Ann. NY Acad. Sci. 1991, 625, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Torok, N.; Krueger, E.; Oswald, B.; McNiven, M.A. Ethanol-induced alterations of the microtubule cytoskeleton in hepatocytes. Am. J. Physiol. 1998, 274, G757–G766. [Google Scholar] [PubMed]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Janke, C. The tubulin code: Molecular components, readout mechanisms, and functions. J. Cell Biol. 2014, 206, 461–472. [Google Scholar] [PubMed]
- Janke, C.; Bulinski, J.C. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.R.; Borisy, G.G. Microtubules are acetylated in domains that turn over slowly. J. Cell Sci. 1989, 92, 57–65. [Google Scholar] [PubMed]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–23. [Google Scholar] [CrossRef] [PubMed]
- Seeley, E.S.; Nachury, M.V. The perennial organelle: Assembly and disassembly of the primary cilium. J. Cell Sci. 2010, 123, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Pous, C.; Chabin, K.; Drechou, A.; Barbot, L.; Phung-Koskas, T.; Settegrana, C.; Bourguet-Kondracki, M.L.; Maurice, M.; Cassio, D.; Guyot, M.; et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. J. Cell Biol. 1998, 142, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Quinones, G.B.; Danowski, B.A.; Devaraj, A.; Singh, V.; Ligon, L.A. The posttranslational modification of tubulin undergoes a switch from detyrosination to acetylation as epithelial cells become polarized. Mol. Biol. Cell 2011, 22, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Zink, S.; Grosse, L.; Freikamp, A.; Banfer, S.; Muksch, F.; Jacob, R. Tubulin detyrosination promotes monolayer formation and apical trafficking in epithelial cells. J. Cell Sci. 2012, 125, 5998–6008. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Tuma, D.J.; Tuma, P.L. Microtubules are more stable and more highly acetylated in ethanol-treated hepatic cells. J. Hepatol. 2006, 44, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, C.S.; Todero, S.L.; McVicker, B.L.; Tuma, P.L.; Sorrell, M.F.; Tuma, D.J. WIF-B cells as a model for alcohol-induced hepatocyte injury. Biochem. Pharmacol. 2004, 67, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.J.; Tuma, D.J.; Tuma, P.L. Hepatic microtubule acetylation and stability induced by chronic alcohol exposure impair nuclear translocation of STAT3 and STAT5B, but not Smad2/3. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1402–G1415. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Black, M.M.; Keyser, P. Acetylation of alpha-tubulin in cultured neurons and the induction of alpha-tubulin acetylation in PC12 cells by treatment with nerve growth factor. J. Neurosci. 1987, 7, 1833–1842. [Google Scholar] [PubMed]
- Brown, A.; Li, Y.; Slaughter, T.; Black, M.M. Composite microtubules of the axon: Quantitative analysis of tyrosinated and acetylated tubulin along individual axonal microtubules. J. Cell Sci. 1993, 104, 339–352. [Google Scholar] [PubMed]
- Cambray-Deakin, M.A.; Burgoyne, R.D. Posttranslational modifications of alpha-tubulin: Acetylated and detyrosinated forms in axons of rat cerebellum. J. Cell Biol. 1987, 104, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Kim, H. Depletion of acetylated alpha-tubulin during microtubule purification from bovine brain gray and white matter regions. J. Neurosci. Res. 1991, 30, 172–182. [Google Scholar] [CrossRef] [PubMed]
- L’Hernault, S.W.; Rosenbaum, J.L. Chlamydomonas alpha-tubulin is posttranslationally modified in the flagella during flagellar assembly. J. Cell Biol. 1983, 97, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Piperno, G.; Fuller, M.T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J. Cell Biol. 1985, 101, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Piperno, G.; LeDizet, M.; Chang, X.J. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J. Cell Biol. 1987, 104, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.W.; Hou, F.; Zhang, J.; Phu, L.; Loktev, A.V.; Kirkpatrick, D.S.; Jackson, P.K.; Zhao, Y.; Zou, H. A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol. Biol. Cell 2011, 22, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Black, M.M. Individual microtubules in the axon consist of domains that differ in both composition and stability. J. Cell Biol. 1990, 111, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Geimer, S.; Teltenkotter, A.; Plessmann, U.; Weber, K.; Lechtreck, K.F. Purification and characterization of basal apparatuses from a flagellate green alga. Cell Motil. Cytoskelet. 1997, 37, 72–85. [Google Scholar] [CrossRef]
- Geuens, G.; Gundersen, G.G.; Nuydens, R.; Cornelissen, F.; Bulinski, J.C.; DeBrabander, M. Ultrastructural colocalization of tyrosinated and detyrosinated alpha-tubulin in interphase and mitotic cells. J. Cell Biol. 1986, 103, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.G.; Bulinski, J.C. Distribution of tyrosinated and nontyrosinated alpha-tubulin during mitosis. J. Cell Biol. 1986, 102, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.G.; Kalnoski, M.H.; Bulinski, J.C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell 1984, 38, 779–789. [Google Scholar] [CrossRef]
- Johnson, K.A. The axonemal microtubules of the Chlamydomonas flagellum differ in tubulin isoform content. J. Cell Sci. 1998, 111, 313–320. [Google Scholar] [PubMed]
- Raybin, D.; Flavin, M. Enzyme which specifically adds tyrosine to the alpha chain of tubulin. Biochemistry 1977, 16, 2189–2194. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.J.; Burgoyne, R.D. Differential localisation of tyrosinated, detyrosinated, and acetylated alpha-tubulins in neurites and growth cones of dorsal root ganglion neurons. Cell Motil. Cytoskelet. 1989, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Paturle-Lafanechere, L.; Edde, B.; Denoulet, P.; van Dorsselaer, A.; Mazarguil, H.; le Caer, J.P.; Wehland, J.; Job, D. Characterization of a major brain tubulin variant which cannot be tyrosinated. Biochemistry 1991, 30, 10523–10528. [Google Scholar] [CrossRef] [PubMed]
- Paturle-Lafanechere, L.; Manier, M.; Trigault, N.; Pirollet, F.; Mazarguil, H.; Job, D. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J. Cell Sci. 1994, 107, 1529–1543. [Google Scholar] [PubMed]
- Rogowski, K.; van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Tort, O.; Tanco, S.; Rocha, C.; Bieche, I.; Seixas, C.; Bosc, C.; Andrieux, A.; Moutin, M.J.; Aviles, F.X.; Lorenzo, J.; et al. The cytosolic carboxypeptidases CCP2 and CCP3 catalyze posttranslational removal of acidic amino acids. Mol. Biol. Cell 2014, 25, 3017–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.E.; Hunt, D.F.; Lee, M.K.; Shabanowitz, J.; Michel, H.; Berlin, S.C.; MacDonald, T.L.; Sundberg, R.J.; Rebhun, L.I.; Frankfurter, A. Characterization of posttranslational modifications in neuron-specific class III beta-tubulin by mass spectrometry. Proc. Natl. Acad. Sci. USA 1991, 88, 4685–4689. [Google Scholar] [CrossRef] [PubMed]
- Audebert, S.; Koulakoff, A.; Berwald-Netter, Y.; Gros, F.; Denoulet, P.; Edde, B. Developmental regulation of polyglutamylated alpha- and beta-tubulin in mouse brain neurons. J. Cell Sci. 1994, 107, 2313–2322. [Google Scholar] [PubMed]
- Bobinnec, Y.; Khodjakov, A.; Mir, L.M.; Rieder, C.L.; Edde, B.; Bornens, M. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J. Cell Biol. 1998, 143, 1575–1589. [Google Scholar] [CrossRef] [PubMed]
- Bre, M.H.; de Nechaud, B.; Wolff, A.; Fleury, A. Glutamylated tubulin probed in ciliates with the monoclonal antibody GT335. Cell Motil. Cytoskelet. 1994, 27, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Edde, B.; Rossier, J.; le Caer, J.P.; Desbruyeres, E.; Gros, F.; Denoulet, P. Posttranslational glutamylation of alpha-tubulin. Science 1990, 247, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, J.P.; Edde, B.; Kann, M.L.; Wolff, A.; Desbruyeres, E.; Denoulet, P. Differential distribution of glutamylated tubulin during spermatogenesis in mammalian testis. Cell Motil. Cytoskelet. 1994, 27, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Mary, J.; Redeker, V.; le Caer, J.P.; Prome, J.C.; Rossier, J. Class I and IVa beta-tubulin isotypes expressed in adult mouse brain are glutamylated. FEBS Lett. 1994, 353, 89–94. [Google Scholar] [CrossRef]
- Mary, J.; Redeker, V.; le Caer, J.P.; Rossier, J.; Schmitter, J.M. Posttranslational modifications in the C-terminal tail of axonemal tubulin from sea urchin sperm. J. Biol. Chem. 1996, 271, 9928–9933. [Google Scholar] [PubMed]
- Mary, J.; Redeker, V.; le Caer, J.P.; Rossier, J.; Schmitter, J.M. Posttranslational modifications of axonemal tubulin. J. Protein Chem. 1997, 16, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, M.; Plessman, U.; Kloppel, K.D.; Wehland, J.; Weber, K. Class II tubulin, the major brain beta tubulin isotype is polyglutamylated on glutamic acid residue 435. FEBS Lett. 1992, 308, 101–105. [Google Scholar] [CrossRef]
- Wolff, A.; de Nechaud, B.; Chillet, D.; Mazarguil, H.; Desbruyeres, E.; Audebert, S.; Edde, B.; Gros, F.; Denoulet, P. Distribution of glutamylated alpha and beta-tubulin in mouse tissues using a specific monoclonal antibody, GT335. Eur. J. Cell Biol. 1992, 59, 425–432. [Google Scholar] [PubMed]
- Bre, M.H.; Redeker, V.; Quibell, M.; Darmanaden-Delorme, J.; Bressac, C.; Cosson, J.; Huitorel, P.; Schmitter, J.M.; Rossler, J.; Johnson, T.; et al. Axonemal tubulin polyglycylation probed with two monoclonal antibodies: Widespread evolutionary distribution, appearance during spermatozoan maturation and possible function in motility. J. Cell Sci. 1996, 109, 727–738. [Google Scholar] [PubMed]
- Levilliers, N.; Fleury, A.; Hill, A.M. Monoclonal and polyclonal antibodies detect a new type of post-translational modification of axonemal tubulin. J. Cell Sci. 1995, 108, 3013–3028. [Google Scholar] [PubMed]
- Redeker, V.; Levilliers, N.; Schmitter, J.M.; le Caer, J.P.; Rossier, J.; Adoutte, A.; Bre, M.H. Polyglycylation of tubulin: A posttranslational modification in axonemal microtubules. Science 1994, 266, 1688–1691. [Google Scholar] [CrossRef] [PubMed]
- Cicchillitti, L.; Penci, R.; di Michele, M.; Filippetti, F.; Rotilio, D.; Donati, M.B.; Scambia, G.; Ferlini, C. Proteomic characterization of cytoskeletal and mitochondrial class III beta-tubulin. Mol. Cancer Ther. 2008, 7, 2070–2079. [Google Scholar] [CrossRef] [PubMed]
- Hino, M.; Kijima-Suda, I.; Nagai, Y.; Hosoya, H. Glycosylation of the alpha and beta tubulin by sialyloligosaccharides. Zool. Sci. 2003, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Kang, J.G.; Park, S.Y.; Lee, J.; Oh, Y.J.; Cho, J.W. O-GlcNAcylation of tubulin inhibits its polymerization. Amino Acids 2011, 40, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J.L.; Vincent, T.S.; Schey, K.L.; Buse, M.G. High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E424–E434. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.M.; Vega, L.R.; Fleming, J.; Bishop, R.; Solomon, F. Single site alpha-tubulin mutation affects astral microtubules and nuclear positioning during anaphase in Saccharomyces cerevisiae: Possible role for palmitoylation of alpha-tubulin. Mol. Biol. Cell 2001, 12, 2672–2687. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Ozols, J.; Caron, J.M. Posttranslational modification of tubulin by palmitoylation: II. Identification of sites of palmitoylation. Mol. Biol. Cell 1997, 8, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Nido, J.; Serrano, L.; Lopez-Otin, C.; Vandekerckhove, J.; Avila, J. Phosphorylation of a neuronal-specific beta-tubulin isotype. J. Biol. Chem. 1990, 265, 13949–13954. [Google Scholar] [PubMed]
- Eipper, B.A. Properties of rat brain tubulin. J. Biol. Chem. 1974, 249, 1407–1416. [Google Scholar] [PubMed]
- Fourest-Lieuvin, A.; Peris, L.; Gache, V.; Garcia-Saez, I.; Juillan-Binard, C.; Lantez, V.; Job, D. Microtubule regulation in mitosis: Tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol. Biol. Cell 2006, 17, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Gard, D.L.; Kirschner, M.W. A polymer-dependent increase in phosphorylation of beta-tubulin accompanies differentiation of a mouse neuroblastoma cell line. J. Cell Biol. 1985, 100, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.E.; Delfino, F.J.; Cheng, H.Y.; Smithgall, T.E. The human C-Fes tyrosine kinase binds tubulin and microtubules through separate domains and promotes microtubule assembly. Mol. Cell Biol. 2004, 24, 9351–9358. [Google Scholar] [CrossRef] [PubMed]
- Panse, V.G.; Hardeland, U.; Werner, T.; Kuster, B.; Hurt, E. A proteome-wide approach identifies sumoylated substrate proteins in yeast. J. Biol. Chem. 2004, 279, 41346–41351. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Acosta, G.; Russell, W.K.; Deyrieux, A.; Russell, D.H.; Wilson, V.G. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol. Cell Proteom. 2005, 4, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Diener, D.R.; Rosenbaum, J.L. The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J. Cell Biol. 2009, 186, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, J.; Feng, J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [PubMed]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Piroli, G.G.; Manuel, A.M.; Walla, M.D.; Jepson, M.J.; Brock, J.W.; Rajesh, M.P.; Tanis, R.M.; Cotham, W.E.; Frizzell, N. Identification of protein succination as a novel modification of tubulin. Biochem. J. 2014, 462, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown—Focus on adducts. Alcohol. Res. Health 2003, 27, 285–290. [Google Scholar] [PubMed]
- Jennett, R.B.; Tuma, D.J.; Sorrell, M.F. Effect of ethanol and its metabolites on microtubule formation. Pharmacology 1980, 21, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Jennett, R.B.; Sorrell, M.F.; Tuma, D.J. Substoichiometric inhibition of microtubule formation by acetaldehyde-tubulin adducts. Biochem. Pharmacol. 1992, 44, 65–72. [Google Scholar] [CrossRef]
- Galligan, J.J.; Smathers, R.L.; Fritz, K.S.; Epperson, L.E.; Hunter, L.E.; Petersen, D.R. Protein carbonylation in a murine model for early alcoholic liver disease. Chem. Res. Toxicol. 2012, 25, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Morinaga, O.; Singh, I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2010, 5, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.J.; Doorn, J.A.; Petersen, D.R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007, 20, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Lowenson, J.D.; Powell, C.D.; Thakur, M.; Bailey, K.; Clarke, S.; Ray, D.E.; Carter, W.G. Proteomic identification of novel substrates of a protein isoaspartyl methyltransferase repair enzyme. J. Biol. Chem. 2006, 281, 32619–32629. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Hood, B.L.; Aragon, R.A.; Hardwick, J.P.; Conrads, T.P.; Veenstra, T.D.; Song, B.J. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics 2006, 6, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.A.; Shepard, B.D.; Kannarkat, G.T.; Rutledge, T.M.; Tuma, D.J.; Tuma, P.L. Microtubule acetylation and stability may explain alcohol-induced alterations in hepatic protein trafficking. Hepatology 2008, 47, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Doody, E.E.; Walker, J.R.; Groebner, J.L.; Tuma, D.J.; Fernandez, D.J.; Tuma, P.L. Ethanol metabolism by CYP2E1 impairs STAT5-mediated growth hormone signaling. 2015; in preparation. [Google Scholar]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an alpha-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+ -dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D.; Joseph, R.A.; Kannarkat, G.T.; Rutledge, T.M.; Tuma, D.J.; Tuma, P.L. Alcohol-induced alterations in hepatic microtubule dynamics can be explained by impaired histone deacetylase 6 function. Hepatology 2008, 48, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Singer, S.J. A possible role for stable microtubules in intracellular transport from the endoplasmic reticulum to the Golgi apparatus. J. Cell Sci. 1994, 107, 1321–1331. [Google Scholar] [PubMed]
- Phung-Koskas, T.; Pilon, A.; Pous, C.; Betzina, C.; Sturm, M.; Bourguet-Kondracki, M.L.; Durand, G.; Drechou, A. STAT5B-mediated growth hormone signaling is organized by highly dynamic microtubules in hepatic cells. J. Biol. Chem. 2005, 280, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D.; Fernandez, D.J.; Tuma, P.L. Alcohol consumption impairs hepatic protein trafficking: Mechanisms and consequences. Genes Nutr. 2009, 5, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.M. The gregarious lipid droplet. J. Biol. Chem. 2008, 283, 28005–28009. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ding, Y.; Chen, Y.; Zhang, S.; Huo, C.; Wang, Y.; Yu, J.; Zhang, P.; Na, H.; Zhang, H.; et al. The proteomics of lipid droplets: Structure, dynamics, and functions of the organelle conserved from bacteria to humans. J. Lipid Res. 2012, 53, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Sahini, N.; Borlak, J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog. Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Andersson, L.; Rutberg, M.; Perman, J.; Lidberg, U.; Johansson, B.R.; Fernandez-Rodriguez, J.; Ericson, J.; Nilsson, T.; Boren, J.; et al. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat. Cell Biol. 2007, 9, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Cuervo, A.M. Autophagy and lipids: Tightening the knot. Semin. Immunopathol. 2010, 32, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Crunk, A.E.; Monks, J.; Murakami, A.; Jackman, M.; Maclean, P.S.; Ladinsky, M.; Bales, E.S.; Cain, S.; Orlicky, D.J.; McManaman, J.L. Dynamic regulation of hepatic lipid droplet properties by diet. PLoS ONE 2013, 8, e67631. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.P.; Welte, M.A.; Block, S.M.; Wieschaus, E.F. Dynein-mediated cargo transport in vivo. A switch controls travel distance. J. Cell Biol. 2000, 148, 945–956. [Google Scholar] [PubMed]
- Shubeita, G.T.; Tran, S.L.; Xu, J.; Vershinin, M.; Cermelli, S.; Cotton, S.L.; Welte, M.A.; Gross, S.P. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell 2008, 135, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Bostrom, P.; Ericson, J.; Rutberg, M.; Magnusson, B.; Marchesan, D.; Ruiz, M.; Asp, L.; Huang, P.; Frohman, M.A.; et al. PLD1 and ERK2 regulate cytosolic lipid droplet formation. J. Cell Sci. 2006, 119, 2246–2257. [Google Scholar] [CrossRef]
- Bostrom, P.; Rutberg, M.; Ericsson, J.; Holmdahl, P.; Andersson, L.; Frohman, M.A.; Boren, J.; Olofsson, S.O. Cytosolic lipid droplets increase in size by microtubule-dependent complex formation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1945–1951. [Google Scholar] [CrossRef] [PubMed]
- Orlicky, D.J.; Monks, J.; Stefanski, A.L.; McManaman, J.L. Dynamics and molecular determinants of cytoplasmic lipid droplet clustering and dispersion. PLoS ONE 2013, 8, e66837. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Guo, X.; Thein, S.; Xu, F.; Sugii, S.; Baas, P.W.; Radda, G.K.; Han, W. Regulation of adipogenesis by cytoskeleton remodelling is facilitated by acetyltransferase MEC-17-dependent acetylation of alpha-tubulin. Biochem. J. 2013, 449, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Mackeh, R.; Lorin, S.; Ratier, A.; Mejdoubi-Charef, N.; Baillet, A.; Bruneel, A.; Hamai, A.; Codogno, P.; Pous, C.; Perdiz, D. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate alpha-tubulin acetyltransferase-1 (alphaTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J. Biol. Chem. 2014, 289, 11816–11828. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978. [Google Scholar]
- You, M.; Rogers, C.Q. Adiponectin: A key adipokine in alcoholic fatty liver. Exp. Biol. Med. 2009, 234, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D.; Tuma, P.L. Alcohol-induced alterations of the hepatocyte cytoskeleton. World J. Gastroenterol. 2010, 16, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Tuma, P.L.; Hubbard, A.L. Transcytosis: Crossing cellular barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [PubMed]
- Akhmanova, A.; Hammer, J.A., 3rd. Linking molecular motors to membrane cargo. Curr. Opin. Cell Biol. 2010, 22, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Kardon, J.R.; Vale, R.D. Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol Cell Biol. 2009, 10, 854–865. [Google Scholar]
- Groebner, J.L.; Fernandez, D.J.; Tuma, D.J.; Tuma, P.L. Alcohol-induced defects in hepatic transcytosis may be explained by impaired dynein function. Mol. Cell Biochem. 2014, 397, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Torok, N.; Marks, D.; Hsiao, K.; Oswald, B.J.; McNiven, M.A. Vesicle movement in rat hepatocytes is reduced by ethanol exposure: Alterations in microtubule-based motor enzymes. Gastroenterology 1997, 113, 1938–1948. [Google Scholar] [CrossRef]
- Shepard, B.D.; Tuma, P.L. Alcohol-induced protein hyperacetylation: Mechanisms and consequences. World J. Gastroenterol. 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Whittaker, M.; Milligan, R.A.; Downing, K.H. High-resolution model of the microtubule. Cell 1999, 96, 79–88. [Google Scholar] [CrossRef]
- Howes, S.C.; Alushin, G.M.; Shida, T.; Nachury, M.V.; Nogales, E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 2014, 25, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Gundersen, G.G. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J. Biol. Chem. 1998, 273, 9797–9803. [Google Scholar] [CrossRef]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.J.; Beranek, V.; Fischermeier, E.; Diez, S. Tubulin acetylation alone does not affect kinesin-1 velocity and run length in vitro. PLoS ONE 2012, 7, e42218. [Google Scholar] [CrossRef] [PubMed]
- Franker, M.A.; Hoogenraad, C.C. Microtubule-based transport—Basic mechanisms, traffic rules and role in neurological pathogenesis. J. Cell Sci. 2013, 126, 2319–2329. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Tischfield, M.A.; Cederquist, G.Y.; Gupta, M.L., Jr.; Engle, E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 2011, 21, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 302, H499–H505. [Google Scholar] [CrossRef] [PubMed]
- Ori-McKenney, K.M.; Xu, J.; Gross, S.P.; Vallee, R.B. A cytoplasmic dynein tail mutation impairs motor processivity. Nat. Cell Biol. 2010, 12, 1228–1234. [Google Scholar] [PubMed]
- Elliott, P.J.; Jirousek, M. Sirtuins: Novel targets for metabolic disease. Curr. Opin. Investig. Drugs 2008, 9, 371–378. [Google Scholar] [PubMed]
- Dekker, F.J.; van den Bosch, T.; Martin, N.I. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov. Today 2014, 19, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Manzo, F.; Tambaro, F.P.; Mai, A.; Altucci, L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin. Ther. Pat. 2009, 19, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Ajmo, J.M.; Liang, X.; Rogers, C.Q.; Pennock, B.; You, M. Resveratrol Alleviates Alcoholic Fatty Liver in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G833–G842. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D.; Tuma, D.J.; Tuma, P.L. Chronic Ethanol Consumption Induces Global Hepatic Protein Hyperacetylation. Alcohol. Clin. Exp. Res. 2010, 34, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Picklo, M.J., Sr. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem. Biophys. Res. Commun. 2008, 21, 615–619. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groebner, J.L.; Tuma, P.L. The Altered Hepatic Tubulin Code in Alcoholic Liver Disease. Biomolecules 2015, 5, 2140-2159. https://doi.org/10.3390/biom5032140
Groebner JL, Tuma PL. The Altered Hepatic Tubulin Code in Alcoholic Liver Disease. Biomolecules. 2015; 5(3):2140-2159. https://doi.org/10.3390/biom5032140
Chicago/Turabian StyleGroebner, Jennifer L., and Pamela L. Tuma. 2015. "The Altered Hepatic Tubulin Code in Alcoholic Liver Disease" Biomolecules 5, no. 3: 2140-2159. https://doi.org/10.3390/biom5032140
APA StyleGroebner, J. L., & Tuma, P. L. (2015). The Altered Hepatic Tubulin Code in Alcoholic Liver Disease. Biomolecules, 5(3), 2140-2159. https://doi.org/10.3390/biom5032140