
Uncommon Glycosidases for the Enzymatic Preparation of Glycosides


Abstract
:1. Introduction
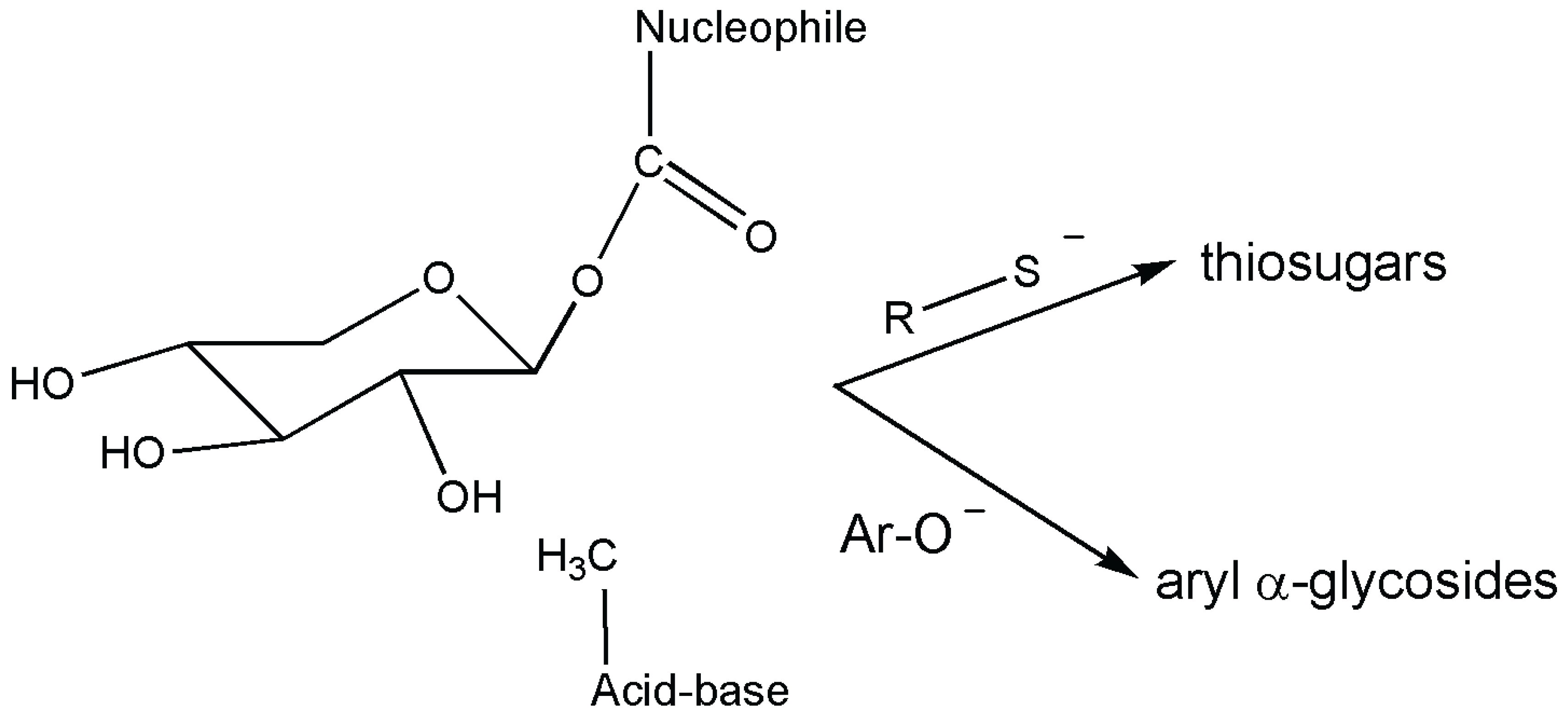
2. Glycosyl Hydrolases: Mechanisms, Use in Synthesis and Rare Examples
3. Arabinofuranosidase



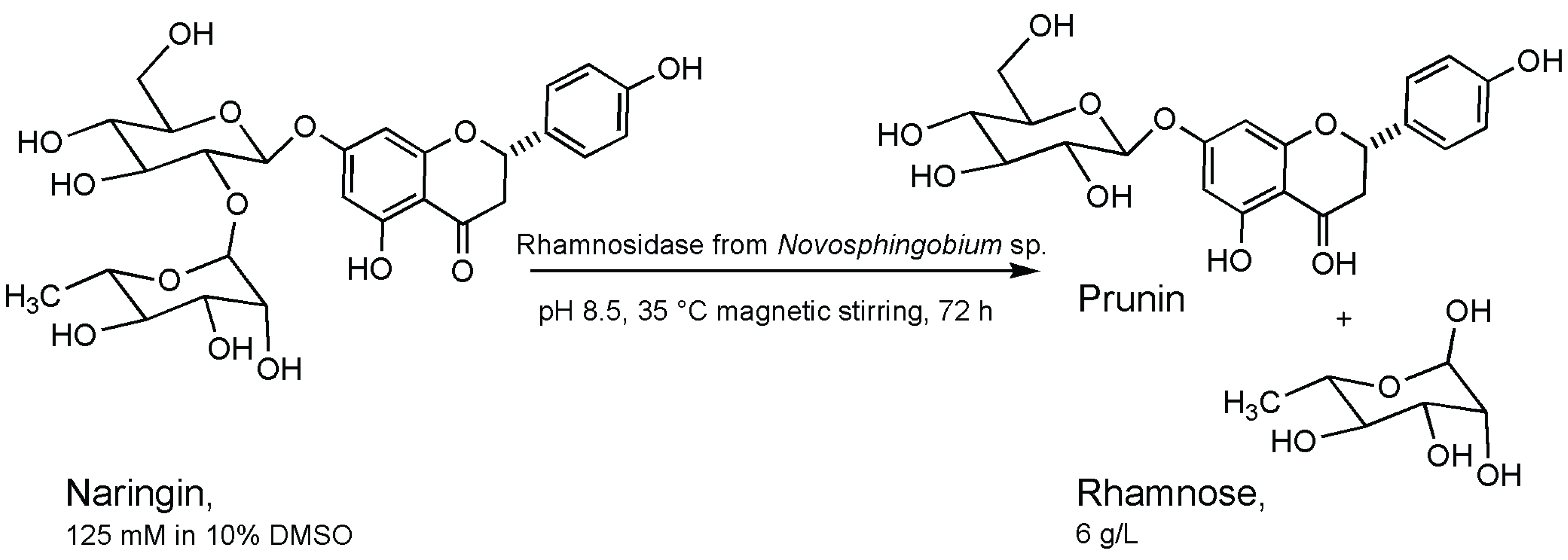
4. Rhamnosidase


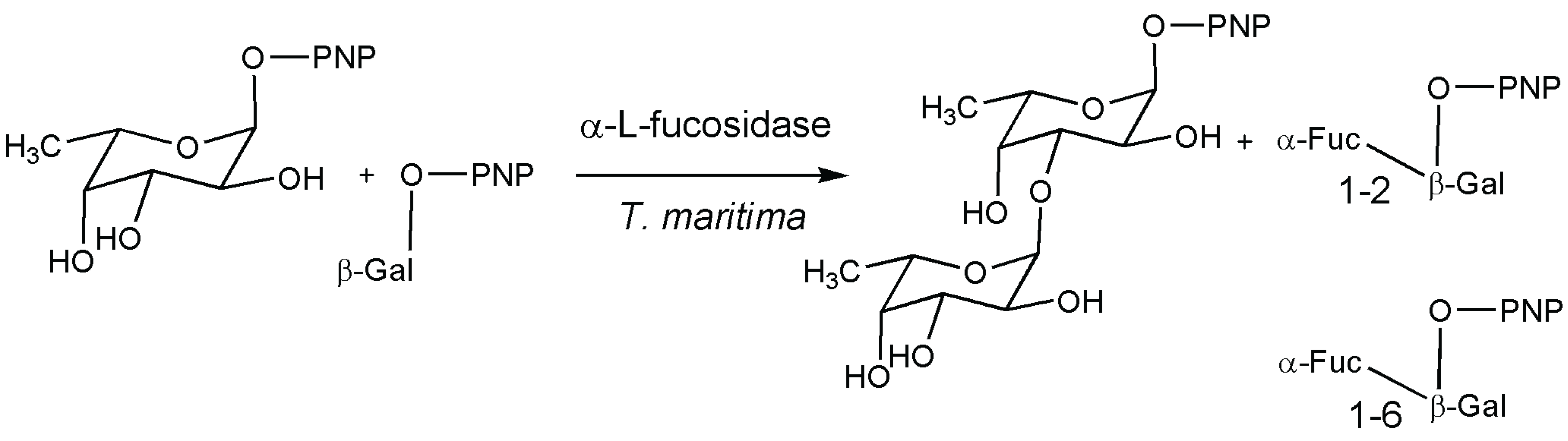
5. Fucosidase

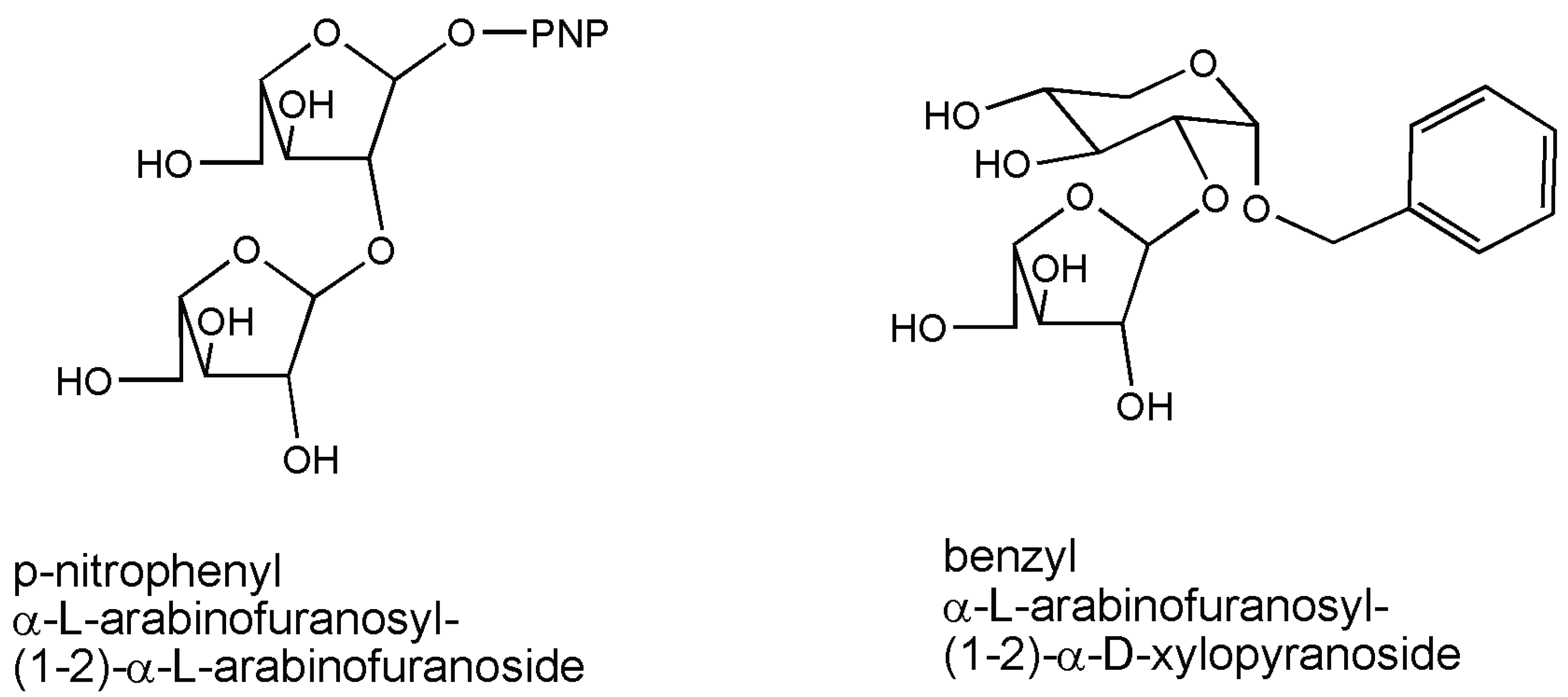{kind=link}
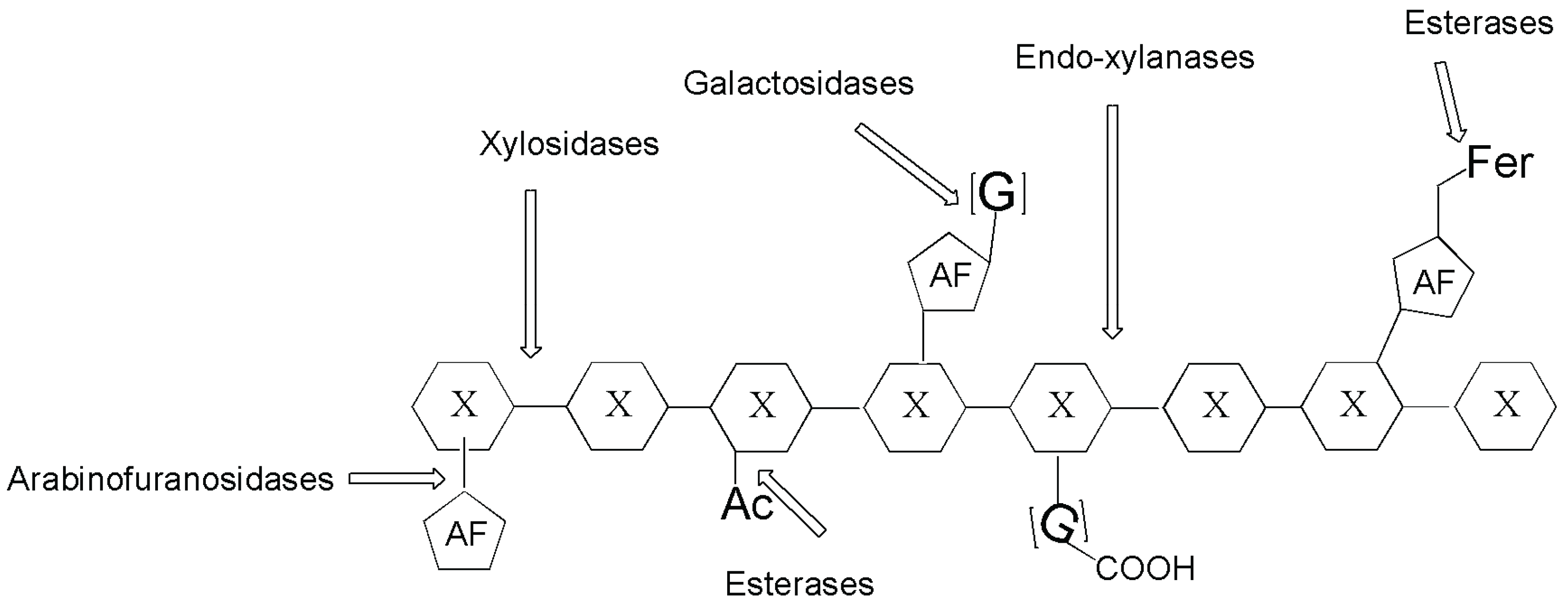{kind=link}
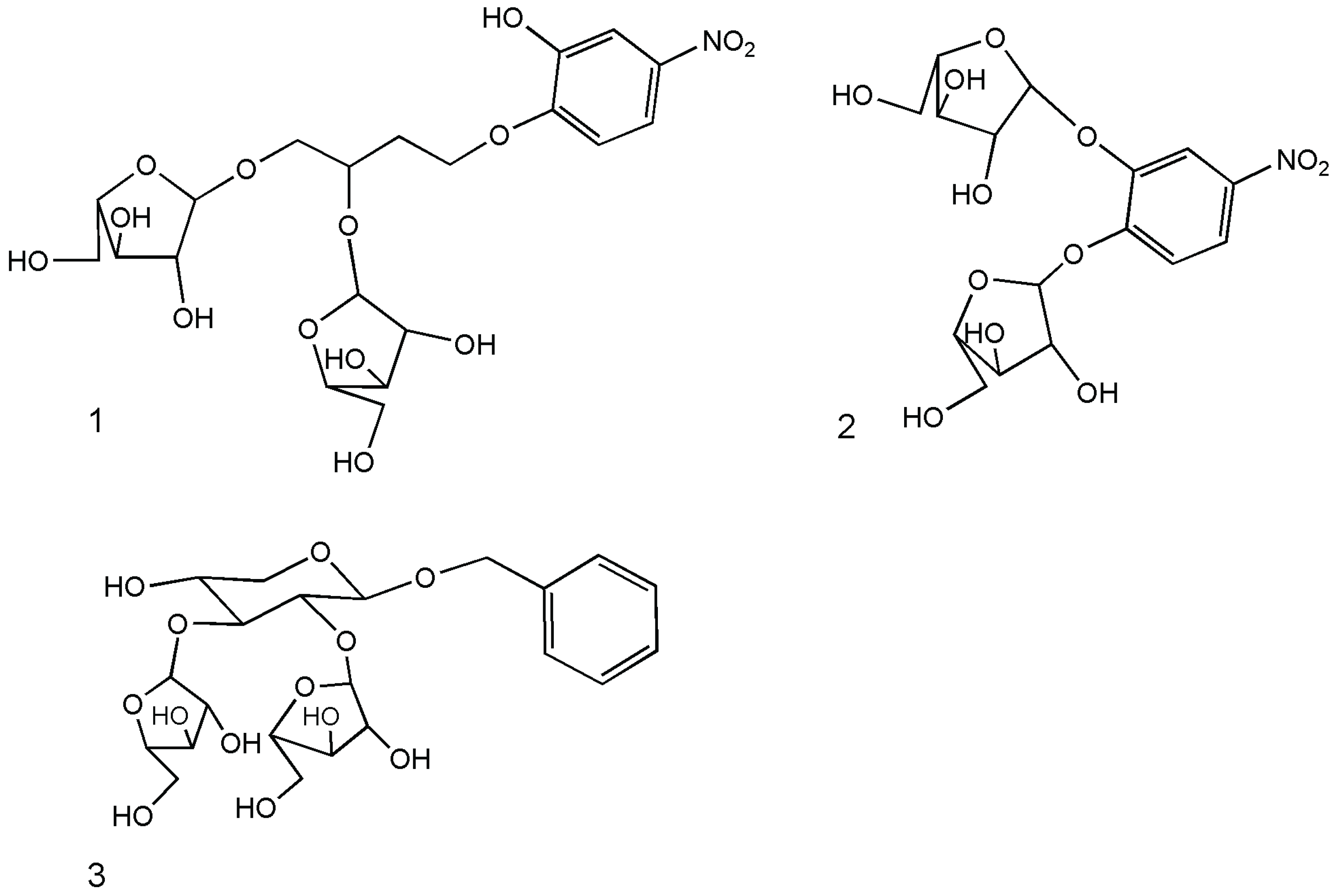{kind=link}
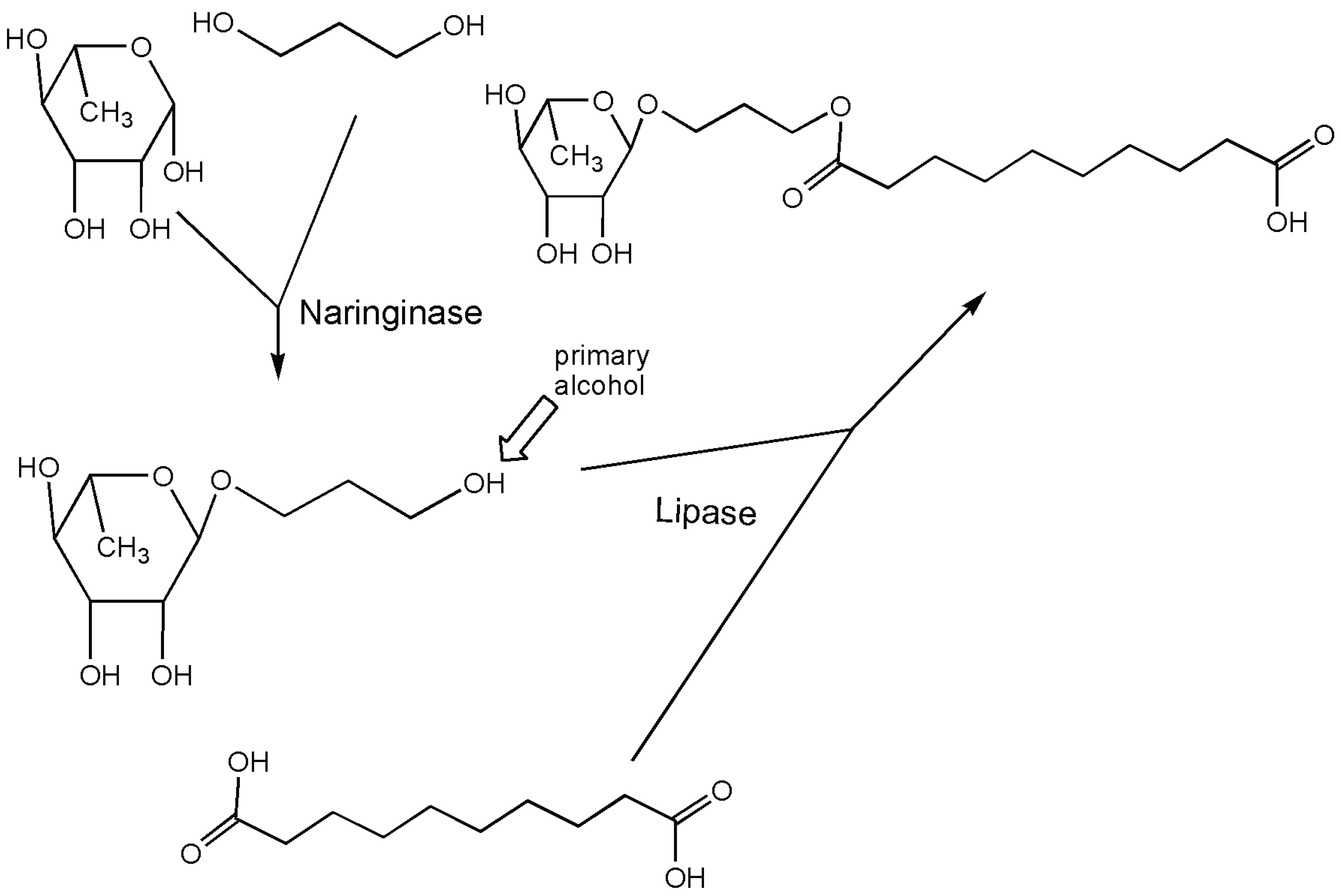{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
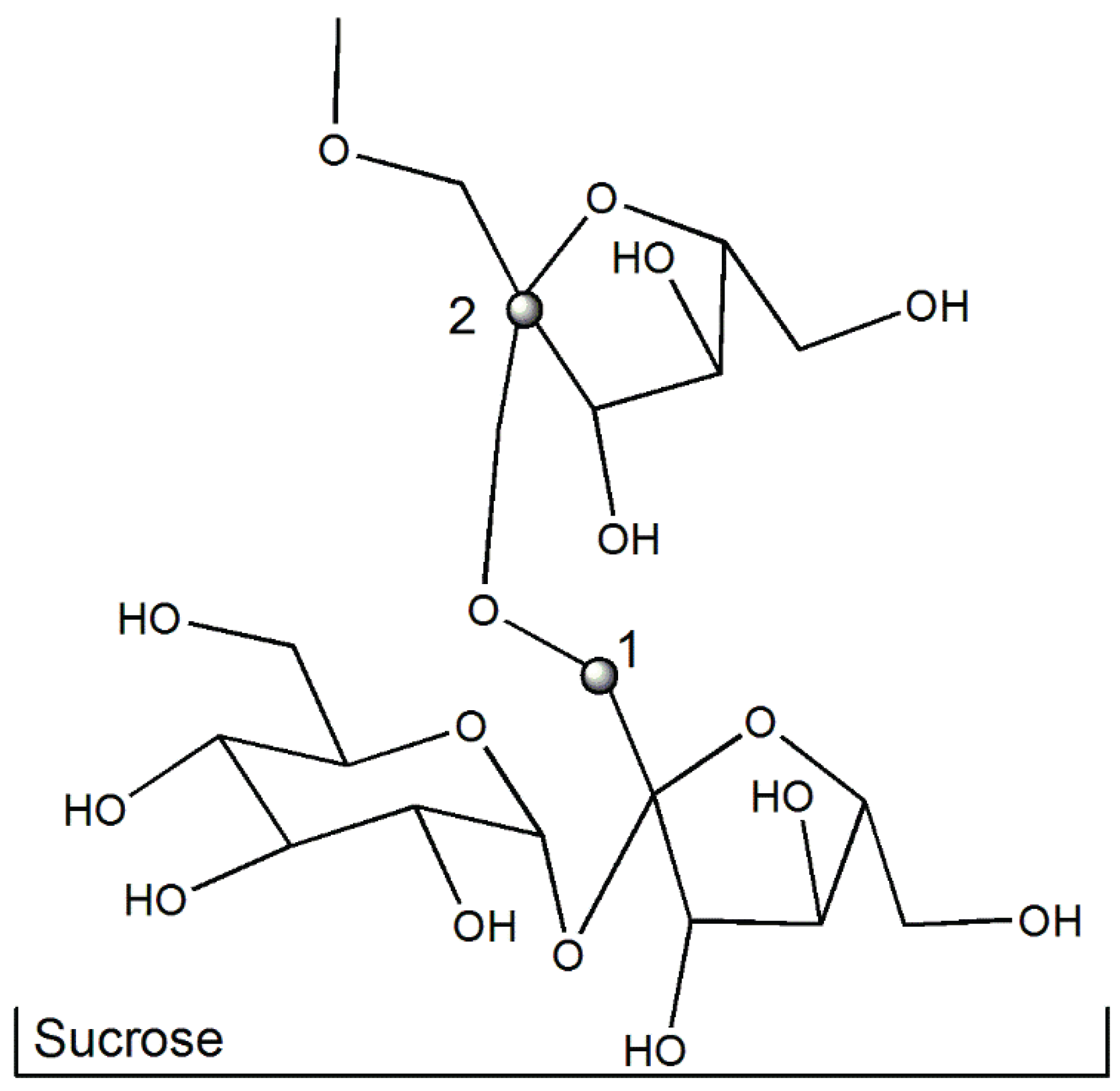{kind=link}
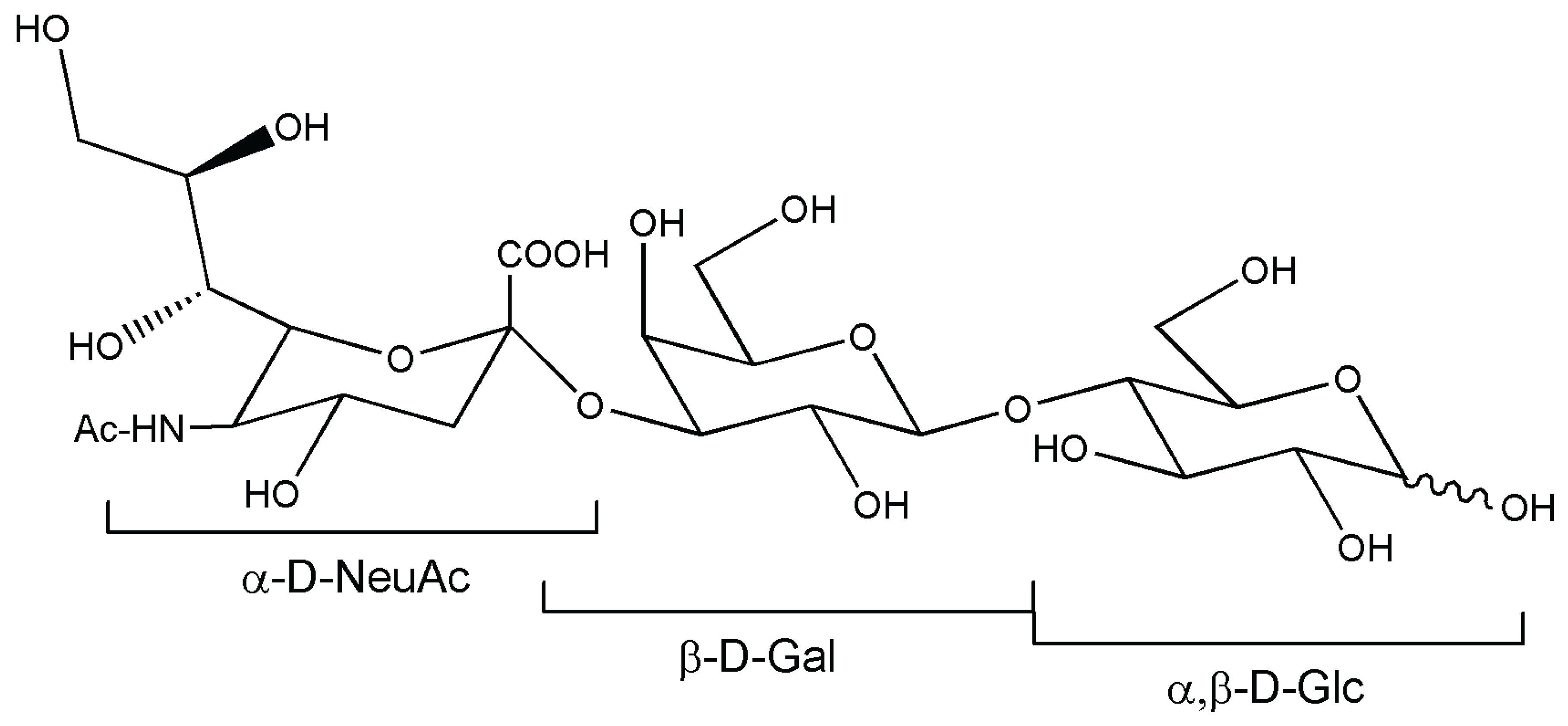{kind=link}
| Enzyme | Products | References |
|---|---|---|
| α-l-fucosidase (porcine liver) | α-l-(1-2)- and α-l-(1-6)- fucosyl derivatives of methyl β-d-Galp; 1,1-trehalose-type disaccharide with Glc-N-Ac. | [45] |
| α-l-fucosidase (porcine liver) | O2, O3, O6 fucosylations of galactose of β-d-Gal-(1-4)-d-GlcNAc. | [46] |
| α-l-fucosidase Aspergillus niger | 3-O-fucosyl disaccharides with 2-acetamido-2-deoxy-d-glucose and glucose. | [47] |
| Fucosidase Corynebacterium sp. and Ampullaria | methyl 2-O-α-l-fucopyranosyl-β-d-galactopyranoside. | [47] |
| Fucosidase Ampullaria | methyl 6-O-α-l-fucopyranosyl-β-d-galactopyranoside | [47] |
| α-l-fucosidase Alcaligenes sp. strain KSF-9687 | O-3-fucosylation of galactose of β-d-Gal-(1-4)-d-GlcNAc; O-3-fucosylation of galactose of lactose; O-3 fucosylation of β-d-Gal-(1-4)-d-GlcNAc-OMe, β-d-Gal-(1-4)-d-Glc-OMe, β-d-Gal-(1-3)-d-Glc-OMe. | [48] |
| α-l-fucosidase Alcaligenes sp. strain | O-3-fucosylation of galactose in PNP-β-lactose and PNP-β-lactosamine. | [49] |
| α-l-fucosidase bovine kidney and testes | O-4-fucosylation of (6-O-Bn)-Glc-NH2-β-SEt. | [50,51] |
| α-l-fucosidase Penicillium multicolor | 3-O-fucosyl disaccharide with 2-acetamido-2-deoxy-d-glucose and methyl or allyl derivatives. | [52] |
| α-l-fucosidase Sulfolobus solfataricus | 3-O and 2-O fucosylation of PNP-β-d-glucoside. | [53] |
| α-l-fucosidase Pecten maximus, canine α-l-fucosidase | highly branched fuco-oligosaccharides as large as tetrasaccharides. | [54] |
| α-l-transfucosidase variants α-l-fucosidase Thermotoga maritima | O-3-fucosylation in self-condensation of PNP-fucoside O-2-fucosylation to PNP-Gal | [55] |

6. Xylanase and Xylosidase

7. Galactosidase
8. Glucuronidase

9. Hyaluronidase
10. Inulinase

11. Sialidase

12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Seeberger, P.H. Automated carbohydrate synthesis as platform to address fundamental aspects of glycobiology-current status and future challenges. Carbohydr. Res. 2008, 343, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Vasu, P.; Persson, S.; Mort, A.J.; Somerville, C.R. Development and application of a suite of polysaccharide-degrading enzymes for analyzing plant cell walls. Proc. Natl. Acad. Sci. USA 2006, 103, 11417–11422. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Henrissat, B. Glycoside hydrolases and glycosyltransferases: Families and functional modules. Curr. Opin. Struct. Biol. 2001, 11, 593–600. [Google Scholar] [CrossRef]
- Trincone, A. Angling for uniqueness in enzymatic preparation of glycosides. Biomolecules 2013, 3, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Monsan, P.; Fauré, R.; O’Donohue, M.J. Glycosynthesis in a waterworld: New insight into the molecular basis of transglycosylation in retaining glycoside hydrolases. Biochem. J. 2015, 467, 17–35. [Google Scholar] [PubMed]
- Wolfenden, R.; Lu, X.; Young, G. Spontaneous hydrolysis of glycosides. J. Am. Chem. Soc. 1998, 120, 6814–6815. [Google Scholar] [CrossRef]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Jongkees, S.A.K.; Withers, S.G. Unusual enzymatic glycoside cleavage mechanisms. Acc. Chem. Res. 2014, 47, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yip, V.L.Y.; Withers, S.G. Mechanisms of enzymatic glycosyl transfer. In Comprehensive Natural Products II Chemistry and Biology; Elsevier: Oxford, UK, 2010; Volume 8, pp. 385–422. [Google Scholar]
- Yip, V.L.Y.; Davis, G.J.; Rajan, S.S.; Yang, X.; Thompson, J.; Anderson, W.F.; Withers, S.G. An unusual mechanism of glycoside hydrolysis involving redox and elimination steps by a family 4 beta-glycosidase from Thermotoga maritima. J. Am. Chem. Soc. 2004, 126, 8354–8355. [Google Scholar] [CrossRef] [PubMed]
- Crout, D.H.; Vic, G. Glycosidases and glycosyl transferases in glycoside and oligosaccharide synthesis. Curr. Opin. Chem. Biol. 1998, 2, 98–111. [Google Scholar] [CrossRef]
- Trincone, A.; Giordano, A. Glycosyl hydrolases and glycosyltransferases in the synthesis of oligosaccharides. Curr. Org. Chem. 2006, 10, 1163–1193. [Google Scholar] [CrossRef]
- Bojarová, P.; Kren, V. Glycosidases: A key to tailored carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, H.; Ge, G.; Zhou, K.; Liu, Y.; Yang, L. Acceptor specificity and transfer efficiency of a β-glycosidase from the Chinese white jade snail. Biosci. Biotechnol. Biochem. 2009, 73, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda, R.H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Rémond, C.; Ferchichi, M.; Aubry, N.; Plantier-Royon, R.; Portella, C.; O'Donohue, M.J. Enzymatic synthesis of alkyl arabinofuranosides using a thermostable α-l-arabinofuranosidase. Tetrahedron Lett. 2002, 43, 9653–9655. [Google Scholar] [CrossRef]
- Rémond, C.; Plantier-Royon, R.; Aubry, N.; Maes, E.; Bliard, C.; O’Donohue, M.J. Synthesis of pentose-containing disaccharides using a thermostable α-l-arabinofuranosidase. Carbohydr. Res. 2004, 339, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Rémond, C.; Plantier-Royon, R.; Aubry, N.; O’Donohue, M.J. An original chemoenzymatic route for the synthesis of β-d-galactofuranosides using an α-l-arabinofuranosidase. Carbohydr. Res. 2005, 340, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Nugier-Chauvin, C.; Rémond, C.; O’Donohue, M.J. Investigation of the specificity of an α-l-arabinofuranosidase using C-2 and C-5 modified α-l-arabinofuranosides. Carbohydr. Res. 2007, 342, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Euzen, R.; Lopez, G.; Nugier-Chauvin, C.; Ferrières, V.; Plusquellec, D.; Rémond, C.; O’Donohue, M.J. A chemoenzymatic approach for the synthesis of unnatural disaccharides containing d-galacto- or d-fucofuranosides. Eur. J. Org. Chem. 2005, 2005, 4860–4869. [Google Scholar] [CrossRef]
- Arab-Jaziri, F.; Bissaro, B.; Tellier, C.; Dion, M.; Fauré, R.; O’Donohue, M.J. Enhancing the chemoenzymatic synthesis of arabinosylated xylo-oligosaccharides by GH51 α-l-arabinofuranosidase. Carbohydr. Res. 2015, 401, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Pennec, A.; Daniellou, R.; Loyer, P.; Nugier-Chauvin, C.; Ferrières, V. Araf51 with improved transglycosylation activities: One engineered biocatalyst for one specific acceptor. Carbohydr. Res. 2015, 402, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissaro, B.; Monsan, P.; Faure, R.; O’Donohue, M.J. Mutation of a pH-modulating residue in a GH51 α-l-arabinofuranosidase leads to a severe reduction of the secondary hydrolysis of transfuranosylation products. Biochim. Biophys. Acta 2014, 1840, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, L.; Ionata, E.; La Cara, F.; Amore, A.; Giacobbe, S.; Pepe, O.; Faraco, V. The effect of Pleurotus ostreatus arabinofuranosidase and its evolved variant in lignocellulosic biomasses conversion. Fungal Genet. Biol. 2014, 72, 162–167. [Google Scholar] [CrossRef]
- Borsenberger, V.; Dornez, E.; Desrousseaux, M.L.; Massou, S.; Tenkanen, M.; Courtin, C.M.; Dumon, C.; O’Donohue, M.J.; Fauré, R. A 1H NMR study of the specificity of α-l-arabinofuranosidases on natural and unnatural substrates. Biochim. Biophys. Acta 2014, 1840, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Guerfali, M.; Gargouri, A.; Belghith, H. Catalytic properties of Talaromyces thermophilus α-l-arabinofuranosidase and its synergistic action with immobilized endo-β-1,4-xylanase. J. Mol. Cat. B Enzym. 2011, 68, 192–199. [Google Scholar] [CrossRef]
- Shi, H.; Li, X.; Gu, H.; Zhang, Y.; Huang, Y.; Wang, L.; Wang, F. Biochemical properties of a novel thermostable and highly xylose-tolerant β-xylosidase/α-arabinosidase from Thermotoga thermarum. Biotechnol. Biofuels 2013. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Saikawa, K.; Kim, S.; Fujita, K.; Ishiwata, A.; Kaeothip, S.; Arakawa, T.; Wakagi, T.; Beckham, G.T.; Ito, Y.; et al. Crystal structure of glycoside hydrolase family 127 β-l-arabinofuranosidase from Bifidobacterium longum. Biochem. Biophys. Res. Commun. 2014, 447, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Takashi, Y.; Obuchi, E.; Kitahara, K.; Suganuma, T. Characterization of a novel β-l-arabinofuranosidase in Bifidobacterium longum: Functional elucidation of a DUF1680 protein family member. J. Biol. Chem. 2014, 289, 5240–5249. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, Y.; Ikeda, K.; Egami, F. α-l-rhamnosidases of the liver of Turbo cornutus and Aspergillus niger. J. Biochem. 1973, 73, 31–37. [Google Scholar] [PubMed]
- Qian, S.; Yu, H.; Zhang, C.; Lu, M.; Wang, H.; Jin, F. Purification and characterization of dioscin-α-l-rhamnosidase from pig liver. Chem. Pharm. Bull. 2005, 53, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. Hydrolysis of flavonoid glycosides by enzymes (rhamnodiastase from Rhamnus and other sources. Arch. Biochem. Biophys. 1962, 99, 476–483. [Google Scholar] [CrossRef]
- Bourbouze, R.; Pratviel-Sosa, F.; Percheron, F. Purification, properties and specificities of a heteroglycosidase from saracen grains. Biochimie 1974, 56, 1305–1313. [Google Scholar] [CrossRef]
- Izzo, V.; Tedesco, P.; Notomista, E.; Pagnotta, E.; Di Donato, A.; Trincone, A.; Tramice, A. α-Rhamnosidase activity in the marine isolate Novosphingobium sp. PP1Y and its use in the bioconversion of flavonoids. J. Mol. Cat. B 2014, 105, 95–103. [Google Scholar] [CrossRef]
- Mazzaferro, L.S.; Piñuel, L.; Erra-Balsells, R.; Giudicessi, S.L.; Breccia, J.D. Transglycosylation specificity of Acremonium sp. α-rhamnosyl-β-glucosidase and its application to the synthesis of the new fluorogenic substrate 4-methylumbelliferyl-rutinoside. Carbohydr. Res. 2012, 347, 69–75. [Google Scholar] [CrossRef] [PubMed]
- De Winter, K.; Šimčíková, D.; Schalck, B.; Weignerová, L.; Pelantova, H.; Soetaert, W.; Desmet, T.; Křen, V. Chemoenzymatic synthesis of α-l-rhamnosides using recombinant α-l-rhamnosidase from Aspergillus terreus. Bioresour. Technol. 2013, 147, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Nott, K.; Richard, G.; Laurent, P.; Jérômed, P.; Blecker, C.; Wathelet, B.; Paquota, M.; Deleua, M. Enzymatic synthesis and surface properties of novel rhamnolipids. Proc. Biochem. 2013, 48, 133–143. [Google Scholar] [CrossRef]
- Martearena, M.R.; Blanco, S.; Ellenrieder, G. Synthesis of alkyl-α-l-rhamnosides by water soluble alcohols enzymatic glycosylation. Bioresour. Technol. 2003, 90, 297–303. [Google Scholar] [CrossRef]
- Puri, M.; Kaur, A.; Schwarz, W.H.; Singh, S.; Kennedy, J.F. Molecular characterization and enzymatic hydrolysis of naringin extracted from kinnow peel waste. Int. J. Biol. Macromol. 2011, 48, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Yadav, P.K.; Yadav, S.; Yadav, K.D.S. α-l-Rhamnosidase: A review. Process Biochem. 2010, 45, 1226–1235. [Google Scholar] [CrossRef]
- Gerstorferová, D.; Fliedrová, B.; Halad, P.; Marhol, P.; Křen, V.; Weignerová, L. Recombinant α-l-rhamnosidase from Aspergillus terreus in selective trimming of rutin. Proc. Biochem. 2012, 47, 828–835. [Google Scholar] [CrossRef]
- Liu, Q.; Lu, L.; Xiao, M. Cell surface engineering of α-l-rhamnosidase for naringin hydrolysis. Bioresour. Technol. 2012, 123, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Perrella, N.N.; Cantinha, R.S.; Nakano, E.; Lopes, A.R. Characterization of α-l-fucosidase and other digestive hydrolases from Biomphalaria glabrata. Acta Trop. 2015, 141, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.J.; Seo, H.C.; Coucke, P.; Tonlorenzi, R.; O’Brien, J.S. Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 1999, 7, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Svennson, S.C.T.; Thiem, J. Purification of α-l-fucosidase by C-glycosylic affinity chromatography, and the enzymic synthesis of α-l-fucosyl disaccharides. Carbohydr. Res. 1990, 200, 391–402. [Google Scholar] [CrossRef]
- Murata, T.; Morimoto, S.; Zeng, X.; Watanabe, S.; Usui, T. Enzymatic synthesis of α-l-fucosyl-N-acetyllactosamines and 3'-O-α-l-fucosyllactose utilizing α-l-fucosidases. Carbohydr. Res. 1999, 320, 192–199. [Google Scholar] [CrossRef]
- Ajisaka, K.; Shirakabe, M. Regioselective synthesis of α-l-fucosyl-containing disaccharides by use of α-l-fucosidases of various origins. Carbohydr. Res. 1992, 224, 291–299. [Google Scholar] [CrossRef]
- Dubey, R.; Reynolds, D.; Abbas, S.A.; Matta, K.L. Synthesis of O-α-l-fucopyranosyl-(1–3)-O-β-d-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-d-glucopyranose-(N-acetyl-3'-O-α-l-fucopyranosyl lactosamine). Carbohydr. Res. 1988, 183, 155–162. [Google Scholar] [CrossRef]
- Zeng, X.; Murata, T.; Usui, T.J. Glycosidase-catalyzed synthesis of fucosyl di- and trisaccharide derivatives using α-l-fucosidase from Alcaligenes sp. Carbohydr. Chem. 2003, 22, 309–316. [Google Scholar] [CrossRef]
- Nilsson, K.G.I.; Eliasson, A.; Larsson-Lorek, U. Production of glucosamine containing disaccharides of the Lewis-A and -x types employing glycosidases. Biotechnol. Lett. 1995, 17, 717–722. [Google Scholar] [CrossRef]
- Nilsson, K.G.I.; Pan, H.; Larsson-Lorek, U. Syntheses of modified carbohydrates with glycosidases: Stereo- and regiospecific syntheses of lactosamine derivatives and related compounds. J. Carbohydr. Chem. 1997, 16, 459–478. [Google Scholar] [CrossRef]
- Ajisaka, K.; Fujimoto, H.; Miyasato, M. An α-l-fucosidase from Penicillium multicolor as a candidate enzyme for the synthesis of (1–3)- linked fucosyl oligosaccharides by transglycosylation. Carbohydr. Res. 1998, 309, 125–129. [Google Scholar] [CrossRef]
- Cobucci-Ponzano, B.; Trincone, A.; Giordano, A.; Rossi, M.; Moracci, M. Identification of the catalytic nucleophile of the family 29 α-l-fucosidase from Sulfolobus solfataricus via chemical rescue of an inactive mutant. Biochemistry 2003, 42, 9525–9531. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O.; Bielicki, J.; Kilonda, A.; Machy, D.; Anson, D.S.; Kenne, L. α-l-fucosidases: Exoglycosidases with unusual transglycosylation properties. Biochemistry 2004, 43, 7881–7891. [Google Scholar] [CrossRef] [PubMed]
- Osanjo, G.; Dion, M.; Drone, J.; Solleux, C.; Tran, V.; Rabiller, C.; Tellier, C. Directed Evolution of the α-l-fucosidase from Thermotoga maritima into an α-l-transfucosidase. Biochemistry 2007, 46, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Roccatagliata, A.J.; Maier, M.S.; Seldes, A.M.; Iorizzi, M.; Minale, L. Starfish saponins. Part II. Steroidal oligoglycosides from the starfish Cosmasterias lurida. J. Nat. Prod. 1994, 57, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Moracci, M.; Ponzano, B.C.; Trincone, A.; Fusco, S.; De Rosa, M.; van der Oost, J.; Sensen, C.W.; Charlebois, R.L.; Rossi, M. Identification and molecular characterization of the first α-xylosidase from an archaeon. J. Biol. Chem. 2000, 275, 22082–22089. [Google Scholar] [CrossRef] [PubMed]
- Trincone, A.; Cobucci Ponzano, B.; Di Lauro, B.; Rossi, M.; Mitsuishi, Y.; Moracci, M. Enzymatic synthesis and hydrolysis of xylogluco-oligosaccharides using the first archaeal α-xylosidase from Sulfolobus solfataricus. Extremophiles 2001, 5, 277–282. [Google Scholar] [CrossRef] [PubMed]
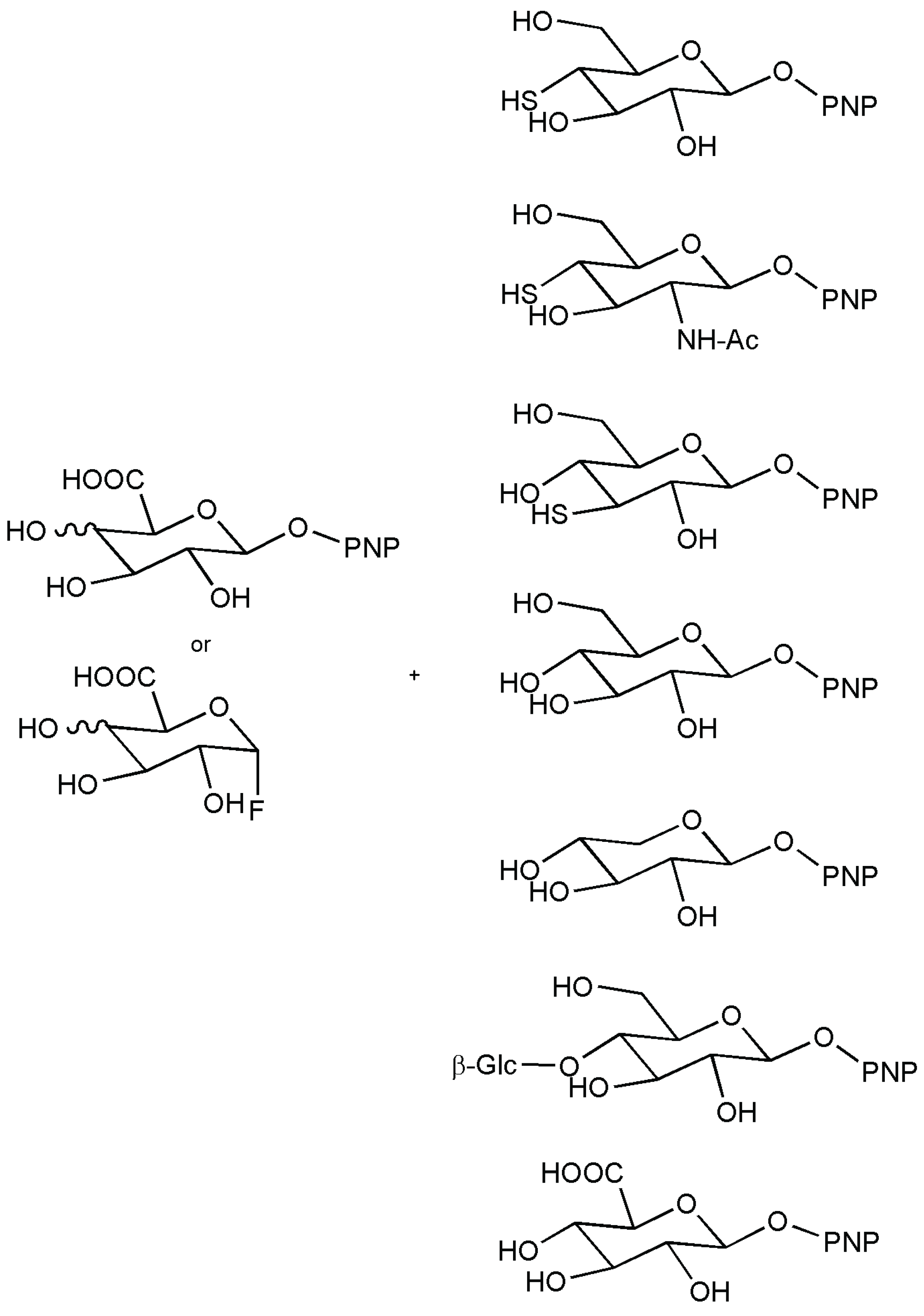
- Li, C.; Kim, J.-H.; Kim, Y.-W. α-Thioglycoligase-based synthesis of O-aryl α-glycosides as chromogenic substrates for α-glycosidases. J. Mol. Cat. B Enzym. 2013, 87, 24–29. [Google Scholar] [CrossRef]
- Dumon, C.; Song, L.; Bozonnet, S.; Fauré, R.; O’Donohue, M.J. Progress and future prospects for pentose-specific biocatalysts in biorefining. Proc. Biochem. 2012, 47, 346–357. [Google Scholar] [CrossRef]
- Faryar, R.; Linares-Pasténa, J.A.; Immerzeel, P.; Mamo, G.; Andersson, M.; Stålbrand, H.; Mattiasson, B.; Nordberg Karlsson, E. Production of prebiotic xylooligosaccharides from alkaline extracted wheat straw using the K80R-variant of a thermostable alkali-tolerant xylanase. Food Bioprod. Process. 2015, 93, 1–10. [Google Scholar] [CrossRef]
- Matsumoto, T.; Shimada, S.; Hata, Y.; Tanaka, T.; Kondo, A. Multi-functional glycoside hydrolase: Blon_0625 from Bifidobacterium longum subsp. infantis ATCC 15697. Enzym. Microb. Technol. 2015, 68, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Lama, L.; Tramice, A.; Finore, I.; Anzelmo, G.; Calandrelli, V.; Pagnotta, E.; Tommonaro, G.; Poli, A.; Di Donato, P.; Nicolaus, B.; et al. Degradative actions of microbial xylanolytic activities on hemicelluloses from rhizome of Arundo donax. AMB Express 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilokpimol, A.; Nakai, H.; Gotfredsen, C.H.; Appeldoorn, M.; Baumann, M.J.; Nakai, N.; Schols, H.A.; Hachem, M.A.; Svensson, B. Enzymatic synthesis of β-xylosyl-oligosaccharides by transxylosylation using two β-xylosidases of glycoside hydrolase family 3 from Aspergillus nidulans FGSC A4. Carbohydr. Res. 2011, 346, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Tramice, A.; Pagnotta, E.; Romano, I.; Gambacorta, A.; Trincone, A. Transglycosylation reactions using glycosyl hydrolases from Thermotoga neapolitana, a marine hydrogen-producing bacterium. J. Mol. Cat. B 2007, 47, 21–27. [Google Scholar] [CrossRef]
- Bakunina, I.Y.; Balabanova, L.A.; Pennacchio, A.; Trincone, A. Hooked on α-d-galactosidases: From biomedicine to enzymatic synthesis. Crit. Rev. Biotechnol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Goulas, T.; Goulas, A.; Tzortzis, G.; Gibson, G.R. A novel α-galactosidase from Bifidobacterium bifidum with transgalactosylating properties: Gene molecular cloning and heterologous expression. Appl. Microbiol. Biotechnol. 2009, 82, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lu, L.; Xiao, M.; Wang, Q.; Lu, Y.; Liu, C.; Wang, P.; Kumagai, H.; Yamamoto, K. Cloning and characterization of a novel α-galactosidase from Bifidobacterium breve 203 capable of synthesizing Gal-α-1,4 linkage. FEMS Microbiol. Lett. 2008, 285, 278–283. [Google Scholar] [CrossRef]
- Simerská, P.; Kuzma, M.; Pisvejcová, A.; Weignerová, L.; Macková, M.; Riva, S.; Kren, V. Application of selectively acylated glycosides for the α-galactosidase-catalyzed synthesis of disaccharides. Folia Microbiol. 2003, 48, 329–337. [Google Scholar] [CrossRef]
- Weignerová, L.; Sedmera, P.; Huňková, Z.; Halada, P.; Křen, V.; Casali, M.; Riva, S. Enzymatic synthesis of iso-globotriose from partially protected lactose. Tetrahedron Lett. 1999, 40, 9297–9299. [Google Scholar] [CrossRef]
- Spangenberg, P.; André, C.; Langlois, V.; Dion, M.; Rabiller, C. α-galactosyl fluoride in transfer reactions mediated by the green coffee beans α-galactosidase in ice. Carbohydr. Res. 2002, 337, 221–228. [Google Scholar] [CrossRef]
- Kurakake, M.; Okumura, T.; Morimoto, Y. Synthesis of galactosyl glycerol from guar gum by transglycosylation of α-galactosidase from Aspergillus sp. MK14. Food Chem. 2015, 172, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Kröger, L.; Mattes, R.; Thiem, J. Transglycosylations employing recombinant α- and β-galactosidases and novel donor substrates. Carbohydr. Res. 2015, 403, 157–166. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.A.; Day, A.J.; Needs, P.W.; Sly, W.S.; O’Brien, N.M.; Williamson, G. Flavonoid glucuronides are substrates for human liver beta-glucuronidase. FEBS Lett. 2001, 503, 103–106. [Google Scholar] [CrossRef]
- Yeşilağaç, R.; Ünak, P.; Medine, E.İ.; İçhedef, Ç.A.; Ertay, T.; Müftüler, F.Z. Enzymatic synthesis of (125/131)I labeled 8-hydroxyquinoline glucuronide and in vitro/in vivo evaluation of biological influence. Appl. Radiat. Isot. 2011, 69, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Salleh, H.M.; Müllegger, J.; Reid, S.P.; Chan, W.Y.; Hwang, J.; Warren, R.A.; Withers, S.G. Cloning and characterization of Thermotoga maritima β-glucuronidase. Carbohydr. Res. 2006, 341, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Müllegger, J.; Chen, H.M.; Chan, W.Y.; Reid, S.P.; Jahn, M.; Warren, R.A.; Salleh, H.M.; Withers, S.G. Thermostable glycosynthases and thioglycoligases derived from Thermotoga maritima β-glucuronidase. Chembiochem 2006, 7, 1028–1030. [Google Scholar] [CrossRef] [PubMed]
- Suresh, C.; Kitaoka, M.; Hayashi, K. A thermostable non-xylanolytic α-glucuronidase of Thermotoga maritima MSB8. Biosci. Biotechnol. Biochem. 2003, 67, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Kakizaki, I.; Suto, S.; Tatara, Y.; Nakamura, T.; Endo, M. Hyaluronan-chondroitin hybrid oligosaccharides as new life science research tools. Biochem. Biophys. Res. Commun. 2012, 423, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Hofinger, E.S.A.; Bernhardt, G.; Buschauer, A. Kinetics of Hyal-1 and PH-20 hyaluronidases: Comparison of minimal substrates and analysis of the transglycosylation reaction. Glycobiology 2007, 17, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, K.; Ishido, K.; Kakizaki, I.; Iwafune, M.; Endo, M. Carriers for enzymatic attachment of glycosaminoglycan chains to peptide. Biochem. Biophys. Res. Commun. 2002, 293, 220–224. [Google Scholar] [CrossRef]
- Madokoro, M.; Ueda, A.; Kiriake, A.; Shiomi, K. Properties and cDNA cloning of a hyaluronidase from the stonefish Synanceia verrucosa venom. Toxicon 2011, 58, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Rigo, D.; Mossi, V.; Golunski, S.; Gde, O.K.; Di Luccio, M.; Dallago, R.; de Oliveira, D.; Oliveira, J.V.; Treichel, H. Enzymatic synthesis of fructooligosaccharides by inulinases from Aspergillus niger and Kluyveromyces marxianus NRRL Y-7571 in aqueous-organic medium. Food Chem. 2013, 138, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.W. Fructooligosaccharides-occurrence, preparation, and application. Enzym. Microb. Technol. 1996, 19, 107–117. [Google Scholar] [CrossRef]
- Tian, F.; Karboune, S.; Hill, A. Synthesis of fructooligosaccharides and oligolevans by the combined use of levansucrase and endo-inulinase in one-step bi-enzymatic system. Innov. Food Sci. Emerg. Technol. 2014, 22, 230–238. [Google Scholar] [CrossRef]
- Tian, F.; Khodadadi, M.; Karboune, S. Optimization of levansucrase/endo-inulinase bi-enzymatic system for the production of fructooligosaccharides and oligolevans from sucrose. J. Mol. Cat. B 2014, 109, 85–93. [Google Scholar] [CrossRef]
- Münger, L.H.; Nyström, L. Enzymatic hydrolysis of steryl glycosides for their analysis in foods. Food Chem. 2014, 163, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Traving, C.; Schauer, R. Structure, function and metabolism of sialic acids. Cell. Mol. Life Sci. 1998, 54, 1330–1349. [Google Scholar] [CrossRef] [PubMed]
- Boons, G.-J.; Demchenko, A.V. Recent Advances in O-sialylation. Chem. Rev. 2000, 100, 4539–4566. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Larsen, D.M.; Jers, C.; Almeida, J.R.M.; Willer, M.; Li, H.; Kirpekar, F.; Kjærulff, L.; Gotfredsen, C.H.; Nordvang, R.T.; et al. Biocatalytic production of 3'-sialyllactose by use of a modified sialidase with superior trans-sialidase activity. Proc. Biochem. 2014, 49, 265–270. [Google Scholar] [CrossRef]
- Zeuner, B.; Luo, J.; Nyffenegger, C.; Aumala, V.; Mikkelsen, J.D.; Meyer, A.S. Optimizing the biocatalytic productivity of an engineered sialidase from Trypanosoma rangeli for 3'-sialyllactose production. Enzym. Microb. Technol. 2014, 55, 85–93. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trincone, A. Uncommon Glycosidases for the Enzymatic Preparation of Glycosides. Biomolecules 2015, 5, 2160-2183. https://doi.org/10.3390/biom5042160
Trincone A. Uncommon Glycosidases for the Enzymatic Preparation of Glycosides. Biomolecules. 2015; 5(4):2160-2183. https://doi.org/10.3390/biom5042160
Chicago/Turabian StyleTrincone, Antonio. 2015. "Uncommon Glycosidases for the Enzymatic Preparation of Glycosides" Biomolecules 5, no. 4: 2160-2183. https://doi.org/10.3390/biom5042160
APA StyleTrincone, A. (2015). Uncommon Glycosidases for the Enzymatic Preparation of Glycosides. Biomolecules, 5(4), 2160-2183. https://doi.org/10.3390/biom5042160