
Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology

{kind=link}
Abstract
:1. Introduction
1.1. The Global Impact of Alcohol Consumption on Human Health
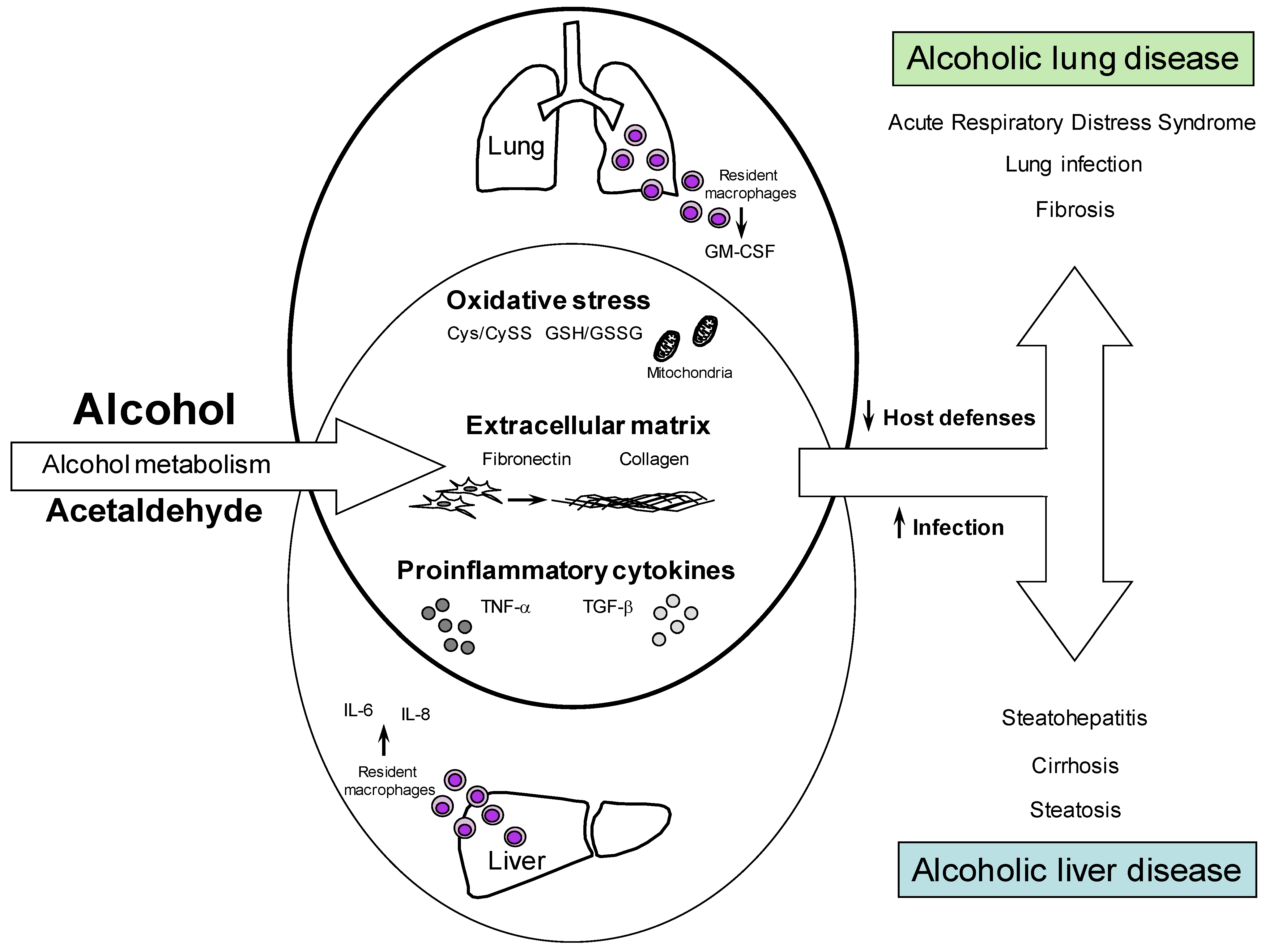
1.2. Alcohol and Liver Diseases
1.3. Alcohol and Lung Disease
1.4. Alcohol Abstinence as a Therapy
1.5. Purpose of This Review
2. Alcoholic Liver Disease-Mechanisms of Injury
2.1. Alcohol and Hepatic Alcohol Metabolism
2.2. Dysregulated Hepatic Inflammation
2.3. Oxidative Stress
2.4. Altered Hepatic Extracellular Matrix Metabolism
3. Alcohol and Mechanisms of Lung Injury
3.1. Alcohol and Pulmonary Alcohol Metabolism
3.2. Dysregulated Pulmonary Inflammation
3.3. Oxidative Stress
3.4. Altered ECM Metabolism
4. Gut:Liver:Lung Axis in Alcohol-Induced Organ Pathology
4.1. Multiorgan Dependencies in Disease
4.2. Alcohol and the Gut:Liver Axis: Historical Perspectives
4.3. Alcohol and the Gut:Lung Axis: Historical Perspectives
4.4. Alcohol and Gut:Liver:Lung Axis: New Areas to Explore

5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- U.S. Department of Health and Human Services. Results from the 2010 National Survey on Drug Use and Health; U.S. Department of Health and Human Services: Washington, DC, USA, 2010.
- World Health Organization. Global Status Report on Alcohol and Health; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Bouchery, E.E.; Harwood, H.J.; Sacks, J.J.; Simon, C.J.; Brewer, R.D. Economic costs of excessive alcohol consumption in the U.S., 2006. Am. J. Prev. Med. 2011, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.F.; Dufour, M.C.; Harford, T.C. Epidemiology of alcoholic liver disease. Semin. Liver Dis. 1988, 8, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Guturu, P.; Hmoud, B.; Kuo, Y.F.; Salameh, H.; Wiesner, R.H. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation 2013, 95, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; McClain, C.J.; Arteel, G.E. Treatment of alcoholic liver disease. Dig. Dis. 2005, 23, 275–284. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, C.; Negri, E.; D’Avanzo, B.; Boyle, P.; Franceschi, S. Medical history and primary liver cancer. Cancer Res. 1990, 50, 6274–6277. [Google Scholar] [PubMed]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Gogel, B.M.; Goldstein, R.M.; Kuhn, J.A.; McCarty, T.M.; Donahoe, A.; Glastad, K. Diagnostic evaluation of hepatocellular carcinoma in a cirrhotic liver. Oncology (Williston Park) 2000, 14, 15–20. [Google Scholar] [PubMed]
- Jong, G.M.; Hsiue, T.R.; Chen, C.R.; Chang, H.Y.; Chen, C.W. Rapidly fatal outcome of bacteremic Klebsiella pneumoniae pneumonia in alcoholics. Chest 1995, 107, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Perlino, C.A.; Rimland, D. Alcoholism, leukopenia, and pneumococcal sepsis. Am. Rev. Respir. Dis. 1985, 132, 757–760. [Google Scholar] [PubMed]
- Cook, R.T. Alcohol abuse, alcoholism, and damage to the immune system—A review. Alcohol. Clin. Exp. Res. 1998, 22, 1927–1942. [Google Scholar]
- Davis, C.C.; Mellencamp, M.A.; Preheim, L.C. A model of pneumococcal pneumonia in chronically intoxicated rats. J. Infect. Dis. 1991, 163, 799–805. [Google Scholar] [CrossRef]
- Lister, P.D.; Gentry, M.J.; Preheim, L.C. Ethanol impairs neutrophil chemotaxis in vitro but not adherence or recruitment to lungs of rats with experimental pneumococcal pneumonia. J. Infect. Dis. 1993, 167, 1131–1137. [Google Scholar] [CrossRef]
- Nelson, S.; Shellito, J.; Mason, C.; Summer, W.R. Alcohol and bacterial pneumonia. Alcohol Health Res. World. 1992, 16, 73–80. [Google Scholar]
- Kershaw, C.D. Alcoholic Lung Disease. Alcohol. Res. Health 2008, 31, 66–75. [Google Scholar] [PubMed]
- Fuxench-Lopez, Z.; Ramirez-Ronda, C.H. Pharyngeal flora in ambulatory alcoholic patients: Prevalence of gram-negative bacilli. Arch. Intern. Med. 1978, 138, 1815–1816. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Gentry-Nielsen, M.J.; Pavlik, J.A.; Sisson, J.H. Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol. Clin. Exp. Res. 2004, 28, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Bagby, G.J.; Xie, M.; Stoltz, D.A.; Summer, W.R.; Nelson, S. Acute ethanol intoxication inhibits neutrophil beta2-integrin expression in rats during endotoxemia. Alcohol. Clin. Exp. Res. 1998, 22, 135–141. [Google Scholar] [CrossRef]
- Bautista, A.P.; Elliott, K.E. Acute ethanol intoxication regulates f-met-leu-phe-induced chemotaxis and superoxide release by neutrophils and Kupffer cells through modulation of the formyl peptide receptor in the rat. Life Sci. 1994, 54, 721–730. [Google Scholar] [CrossRef]
- MacGregor, R.R. In vivo neutrophil delivery in men with alcoholic cirrhosis is normal despite depressed in vitro chemotaxis. Alcohol. Clin. Exp. Res. 1990, 14, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Keshavarzian, A.; Kottapalli, V.; Badie, B.; Winship, D.; Fields, J.Z. Human neutrophil functions are inhibited in vitro by clinically relevant ethanol concentrations. Alcohol. Clin. Exp. Res. 1996, 20, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Bucher, B.; Moore, F.A.; Moore, E.E.; Parsons, P.E. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA 1996, 275, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. J. Am. Med. Assoc. 2012, 307, 2526–2533. [Google Scholar]
- Moss, M.; Burnham, E.L. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit. Care Med. 2003, 31, S207–S212. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.O.; Jensen, J.S.; Ritzenthaler, J.D.; Roman, J.; Pelaez, A.; Guidot, D.M. Alcohol primes the airway for increased interleukin-13 signaling. Alcohol. Clin. Exp. Res. 2009, 33, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Applewhite, L.; Ritzenthaler, J.D.; Roman, J.; Fernandez, A.L.; Eaton, D.C.; Brown, L.A.; Guidot, D.M. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J. Immunol. 2005, 175, 6837–6845. [Google Scholar] [CrossRef]
- Roman, J.; Ritzenthaler, J.D.; Bechara, R.; Brown, L.A.; Guidot, D. Ethanol stimulates the expression of fibronectin in lung fibroblasts via kinase-dependent signals that activate CREB. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L975–L987. [Google Scholar] [CrossRef]
- Lois, M.; Brown, L.A.; Moss, I.M.; Roman, J.; Guidot, D.M. Ethanol ingestion increases activation of matrix metalloproteinases in rat lungs during acute endotoxemia. Am. J. Respir. Crit. Care Med. 1999, 160, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Holguin, F.; Moss, I.; Brown, L.A.; Guidot, D.M. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J. Clin. Investig. 1998, 101, 761–768. [Google Scholar] [CrossRef]
- Otis, J.S.; Brown, L.A.; Guidot, D.M. Oxidant-induced atrogin-1 and transforming growth factor-beta1 precede alcohol-related myopathy in rats. Muscle Nerve 2007, 36, 842–848. [Google Scholar] [CrossRef]
- Guidot, D.M.; Roman, J. Chronic ethanol ingestion increases susceptibility to acute lung injury: Role of oxidative stress and tissue remodeling. Chest 2002, 122, 309S–314S. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.J., Jr.; Klatskin, G. Duration of survival in patients with Laennec’s cirrhosis. Influence of alcohol withdrawal, and possible effects of recent changes in general management of the disease. Am. J. Med. 1968, 44, 406–420. [Google Scholar] [CrossRef]
- Moss, M.; Guidot, D.M.; Wong-Lambertina, M.; Ten Hoor, T.; Perez, R.L.; Brown, L.A. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am. J. Respir. Crit. Care Med. 2000, 161, 414–419. [Google Scholar] [CrossRef]
- Arteel, G.E. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 2003, 124, 778–790. [Google Scholar] [CrossRef]
- Deaciuc, I.V.; Nikolova-Karakashian, M.; Fortunato, F.; Lee, E.Y.; Hill, D.B.; McClain, C.J. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol. Clin. Exp. Res. 2000, 24, 1557–1565. [Google Scholar] [CrossRef]
- Beier, J.I.; Luyendyk, J.P.; Guo, L.; von Montfort, C.; Staunton, D.E.; Arteel, G.E. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology 2009, 49, 1545–1553. [Google Scholar] [CrossRef]
- Ontko, J.A. Effects of ethanol on the metabolism of free fatty acids in isolated liver cells. J. Lipid Res. 1973, 14, 78–86. [Google Scholar] [PubMed]
- Lieber, C.S. Metabolic effects of acetaldehyde. Biochem. Soc. Trans. 1988, 16, 241–247. [Google Scholar] [CrossRef]
- Klassen, L.W.; Tuma, D.; Sorrell, M.F. Immune mechanisms of alcohol-induced liver disease. Hepatology 1995, 22, 355–357. [Google Scholar]
- Niemela, O. Distribution of ethanol-induced protein adducts in vivo: Relationship to tissue injury. Free Rad. Biolol. Med. 2001, 31, 1533–1538. [Google Scholar] [CrossRef]
- Thiele, G.M.; Worrall, S.; Tuma, D.J.; Klassen, L.W.; Wyatt, T.A.; Nagata, N. The chemistry and biological effects of malondialdehyde-acetaldehyde adducts. Alcohol. Clin. Exp. Res. 2001, 25, 218S–224S. [Google Scholar] [CrossRef]
- Visvanathan, K.; Crum, R.M.; Strickland, P.T.; You, X.; Ruczinski, I.; Berndt, S.I.; Alberg, A.J.; Hoffman, S.C.; Comstock, G.W.; Bell, D.A.; et al. Alcohol dehydrogenase genetic polymorphisms, low-to-moderate alcohol consumption, and risk of breast cancer. Alcohol. Clin. Exp. Res. 2007, 31, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, C.J. The role of acetaldehyde in the actions of alcohol (update 2000). Alcohol. Clin. Exp. Res. 2001, 25, 15S–32S. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta 1997, 257, 59–84. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Yuan, Q.X.; Li, J.; French, B.A.; Fang, C.; Ingelman-Sundberg, M.; French, S.W. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem. Biophys. Res. Commun. 2000, 279, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, G.; Ingelman-Sundberg, M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem. Pharmacol. 1989, 38, 1313–1319. [Google Scholar] [CrossRef]
- Enomoto, N.; Ikejima, K.; Bradford, B.U.; Rivera, C.A.; Kono, H.; Brenner, D.A.; Thurman, R.G. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 1998, 115, 443–451. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Thurman, R.G. Up-regulation of CD14 in liver caused by acute ethanol involves oxidant-dependent AP-1 pathway. J. Biol. Chem. 2003, 278, 8435–8441. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol 2004, 33, 147–155. [Google Scholar] [CrossRef]
- Goral, J.; Kovacs, E.J. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J. Immunol. 2005, 174, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.; Rubin, K.P.; Lieber, C.S. Depressed hepatic glutathione and increased diene conjugates in alcoholic liver disease: Evidence of lipid peroxidation. Dig. Dis. Sci. 1983, 28, 585–589. [Google Scholar]
- Bigatello, L.M.; Broitman, S.A.; Fattori, L.; di Paoli, M.; Pontello, M.; Bevilacqua, G.; Nespoli, A. Endotoxemia, encephalopathy, and mortality in cirrhotic patients. Am. J. Gastroenterol. 1987, 82, 11–15. [Google Scholar]
- Deaciuc, I.V.; Fortunato, F.; D’Souza, N.B.; Hill, D.B.; Schmidt, J.; Lee, E.Y.; McClain, C.J. Modulation of caspase-3 activity and Fas ligand mRNA expression in rat liver cells in vivo by alcohol and lipopolysaccharide. Alcohol. Clin. Exp. Res. 1999, 23, 349–356. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Cohen, D.A. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 1989, 9, 349–351. [Google Scholar] [CrossRef]
- Hill, D.B.; Marsano, L.; Cohen, D.; Allen, J.; Shedlofsky, S.; McClain, C.J. Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J. Lab. Clin. Med. 1992, 119, 547–552. [Google Scholar] [CrossRef]
- Hill, D.B.; Marsano, L.S.; McClain, C.J. Increased plasma interleukin-8 concentrations in alcoholic hepatitis. Hepatology 1993, 18, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja, M.N.; McClain, C.J.; Barve, S.; Vaddi, K.; Hill, D.B. Increased monocyte MCP-1 production in acute alcoholic hepatitis. Cytokine 1999, 11, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Fisher, N.C.; Neil, D.A.; Williams, A.; Adams, D.H. Serum concentrations and peripheral secretion of the beta chemokines monocyte chemoattractant protein 1 and macrophage inflammatory protein 1alpha in alcoholic liver disease. Gut 1999, 45, 416–420. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Liu, H.; Jones, B.E.; Bradham, C.; Czaja, M.J. Increased cytochrome P-450 2E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G257–G266. [Google Scholar] [CrossRef]
- Di Luzio, N.R. A mechanism of the acute ethanol-induced fatty liver and the modification of liver injury by antioxidants. Lab. Investig. 1966, 15, 50–63. [Google Scholar]
- Shaw, S.; Jayatilleke, E.; Ross, W.A.; Gordon, E.R.; Lieber, S. Ethanol-induced lipid peroxidation: Potentiation by long-term alcohol feeding and attenuation by methionine. J. Lab. Clin. Med. 1981, 98, 417–424. [Google Scholar]
- Patek, A.J., Jr. Alcohol, malnutrition, and alcoholic cirrhosis. Am. J. Clin. Nutr. 1979, 32, 1304–1312. [Google Scholar] [PubMed]
- Bujanda, L. The effects of alcohol consumption upon the gastrointestinal tract. Am. J. Gastroenterol. 2000, 95, 3374–3382. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. ALCOHOL: Its metabolism and interaction with nutrients. Annu. Rev. Nutr. 2000, 20, 395–430. [Google Scholar] [CrossRef]
- Kono, H.; Arteel, G.E.; Rusyn, I.; Sies, H.; Thurman, R.G. Ebselen prevents early alcohol-induced liver injury in rats. Free Radic. Biolol. Med. 2001, 30, 403–411. [Google Scholar] [CrossRef]
- Kono, H.; Rusyn, I.; Uesugi, T.; Yamashina, S.; Connor, H.D.; Dikalova, A.; Mason, R.P.; Thurman, R.G. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1005–G1012. [Google Scholar] [PubMed]
- Kono, H.; Rusyn, I.; Connor, H.; Mason, R.P.; Thurman, R.G. Allopurinol prevents early alcohol-induced liver injury in rats. J. Pharmacol. Exp. Ther. 2000, 293, 296–303. [Google Scholar] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Pietsch, E.C.; Cunningham, C.C. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic. Biol. Med. 1999, 27, 891–900. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [PubMed]
- Kono, H.; Uesugi, T.; Froh, M.; Rusyn, I.; Bradford, B.U.; Thurman, R.G. ICAM-1 is involved in the mechanism of alcohol-induced liver injury: Studies with knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1289–G1295. [Google Scholar] [PubMed]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 2002, 36, 200–209. [Google Scholar] [CrossRef]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Kirby, J.A.; Yip, W.W.; Jones, D.E.; Burt, A.D. Biliary epithelial-mesenchymal transition in posttransplantation recurrence of primary biliary cirrhosis. Hepatology 2007, 45, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; McHutchison, J.; Manns, M.; Trepo, C.; Lindsay, K.; Goodman, Z.; Ling, M.H.; Albrecht, J. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002, 122, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R.; Thomson, N.M.; Glasgow, E.F.; Atkins, R.C. The effect of defibrination on macrophage participation in rabbit nephrotoxic nephritis: Studies using glomerular culture and electronmicroscopy. Clin. Exp. Immunol. 1979, 37, 38–43. [Google Scholar] [PubMed]
- Loike, J.D.; el Khoury, J.; Cao, L.; Richards, C.P.; Rascoff, H.; Mandeville, J.T.; Maxfield, F.R.; Silverstein, S.C. Fibrin regulates neutrophil migration in response to interleukin 8, leukotriene B4, tumor necrosis factor, and formyl-methionyl-leucyl-phenylalanine. J. Exp. Med. 1995, 181, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Ganey, P.E.; Luyendyk, J.P.; Maddox, J.F.; Roth, R.A. Adverse hepatic drug reactions: Inflammatory episodes as consequence and contributor. Chem. Biol. Interact. 2004, 150, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.M.; Schultze, A.E.; Schwartz, K.A.; Scott, M.A.; Davis, J.M.; Roth, R.A. The thrombin inhibitor, hirudin, attenuates lipopolysaccharide-induced liver injury in the rat. J. Pharmacol. Exp. Ther. 1996, 278, 378–383. [Google Scholar] [PubMed]
- Hodivala-Dilke, K.M.; Reynolds, A.R.; Reynolds, L.E. Integrins in angiogenesis: Multitalented molecules in a balancing act. Cell Tissue Res. 2003, 314, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.F.; Chan, H.W.; Wickline, S.A.; Lanza, G.M.; Pham, C.T. Alphavbeta3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J. 2009, 23, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Manautou, J.E.; Buss, N.J.; Carlson, G.P. Oxidative and non-oxidative metabolism of ethanol by the rabbit lung. Toxicol. Lett. 1992, 62, 93–99. [Google Scholar] [CrossRef]
- George, S.C.; Hlastala, M.P.; Souders, J.E.; Babb, A.L. Gas exchange in the airways. J. Aerosol. Med. 1996, 9, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hlastala, M.P. The alcohol breath test—A review. J. Appl. Physiol. 1998, 84, 401–408. [Google Scholar] [PubMed]
- Masoro, E.J.; Abramovitch, H.; Birchard, J.R. Metabolism of C14 ethanol by surviving rat tissues. Am. J. Physiol. 1953, 173, 37–40. [Google Scholar] [PubMed]
- Freund, G.; O’Hollaren, P. Acetaldehyde concentrations in alveolar air following a standard dose of ethanol in man. J. Lipid Res. 1965, 6, 471–477. [Google Scholar]
- Karkoulias, K.; Tsitsaras, H.; Patouchas, D.; Sampsonas, F.; Likouras, D.; Kaparianos, A.; Spiropoulos, K. The alcoholic lung disease: Historical background and clinical features. Medicina (Kaunas) 2008, 44, 651–664. [Google Scholar] [PubMed]
- Majno, G.J. Cells, Tissues and Disease, 2nd ed.; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Kumar, V.C.; Robbins, S.L. Robbins Basic Pathology, 7th ed.; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Omidvari, K.; Casey, R.; Nelson, S.; Olariu, R.; Shellito, J.E. Alveolar macrophage release of tumor necrosis factor-alpha in chronic alcoholics without liver disease. Alcohol. Clin. Exp. Res. 1998, 22, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.S.; Xie, J.; Wang, Y.; Kolls, J.; Malinski, T.; Summer, W.R.; Nelson, S. Ethanol suppresses LPS-induced mRNA for nitric oxide synthase II in alveolar macrophages in vivo and in vitro. Alcohol 1994, 11, 539–547. [Google Scholar] [CrossRef]
- Standiford, T.J.; Danforth, J.M. Ethanol feeding inhibits proinflammatory cytokine expression from murine alveolar macrophages ex vivo. Alcohol. Clin. Exp. Res. 1997, 21, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, B.C.; Whitsett, J.A. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 2002, 64, 775–802. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, A.; Bechara, R.I.; Joshi, P.C.; Brown, L.A.; Guidot, D.M. Granulocyte/macrophage colony-stimulating factor treatment improves alveolar epithelial barrier function in alcoholic rat lung. Am. J. Physiol. 2004, 286, L106–L111. [Google Scholar] [CrossRef]
- Joshi, P.C.; Mehta, A.; Jabber, W.S.; Fan, X.; Guidot, D.M. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am. J. Physiol. Cell Mol. Biol. 2009, 41, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.J.; Joshi, P.C.; Fan, X.; Brown, L.A.; Ritzenthaler, J.D.; Roman, J.; Guidot, D.M. Zinc supplementation restores PU.1 and Nrf2 nuclear binding in alveolar macrophages and improves redox balance and bacterial clearance in the lungs of alcohol-fed rats. Alcohol. Clin. Exp. Res. 2011, 35, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Kurts, C. Infiltrating monocytes versus resident Kupffer cells: Do alternatively activated macrophages need to be targeted alternatively? Hepatology 2011, 54, 2267–2270. [Google Scholar] [CrossRef]
- De Rycke, L.; Baeten, D.; Foell, D.; Kruithof, E.; Veys, E.M.; Roth, J.; de Keyser, F. Differential expression and response to anti-TNFalpha treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J. Pathol. 2005, 206, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Astry, C.L.; Warr, G.A.; Jakab, G.J. Impairment of polymorphonuclear leukocyte immigration as a mechanism of alcohol-induced suppression of pulmonary antibacterial defenses. Am. Rev. Respirat. Dis. 1983, 128, 113–117. [Google Scholar]
- Balamayooran, G.; Batra, S.; Fessler, M.B.; Happel, K.I.; Jeyaseelan, S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am. J. Respirat. Cell Mol. Biol. 2010, 43, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.J.; Marrero, L.; Song, K.; Oliver, P.; Chin, S.Y.; Simon, H.; Schurr, J.R.; Zhang, Z.; Thoppil, D.; Lee, S.; et al. Acute alcohol inhibits TNF-alpha processing in human monocytes by inhibiting TNF/TNF-alpha-converting enzyme interactions in the cell membrane. J. Immunol. 2003, 170, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.L.; Koval, M.; Fan, X.; Guidot, D.M. Chronic alcohol ingestion alters claudin expression in the alveolar epithelium of rats. Alcohol 2007, 41, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bechara, R.I.; Brown, L.A.; Roman, J.; Joshi, P.C.; Guidot, D.M. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am. J. Respir. Crit. Care Med. 2004, 170, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Bechara, R.I.; Pelaez, A.; Palacio, A.; Joshi, P.C.; Hart, C.M.; Brown, L.A.; Raynor, R.; Guidot, D.M. Angiotensin II mediates glutathione depletion, transforming growth factor-beta1 expression, and epithelial barrier dysfunction in the alcoholic rat lung. Am. J. Physiol. 2005, 289, L363–L370. [Google Scholar]
- Sheppard, D. Modulation of acute lung injury by integrins. Proc. Am. Thorac. Soc. 2012, 9, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Pittet, J.F.; Griffiths, M.J.; Geiser, T.; Kaminski, N.; Dalton, S.L.; Huang, X.; Brown, L.A.; Gotwals, P.J.; Koteliansky, V.E.; Matthay, M.A.; et al. TGF-beta is a critical mediator of acute lung injury. J. Clin. Investig. 2001, 107, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. (Ed.) Oxidative stress: Introductory remarks. In Oxidative Stress; Academic Press: London, UK, 1985; pp. 1–8.
- Brown, L.A.; Harris, F.L.; Bechara, R.; Guidot, D.M. Effect of chronic ethanol ingestion on alveolar type II cell: Glutathione and inflammatory mediator-induced apoptosis. Alcohol. Clin. Exp. Res. 2001, 25, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Guidot, D.M.; Modelska, K.; Lois, M.; Jain, L.; Moss, I.M.; Pittet, J.F.; Brown, L.A. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am. J. Physiol. 2000, 279, L127–L135. [Google Scholar]
- Guidot, D.M.; Brown, L.A. Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed rats. Alcohol. Clin. Exp. Res. 2000, 24, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Ping, X.D.; Harris, F.L.; Gauthier, T.W. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L824–L832. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Harris, F.L.; Ping, X.D.; Gauthier, T.W. Chronic ethanol ingestion and the risk of acute lung injury: A role for glutathione availability? Alcohol 2004, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.Y.; Burnham, E.L.; Moss, M.; Brown, L.A. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am. J. Respir. Crit. Care Med. 2007, 176, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Polikandriotis, J.A.; Rupnow, H.L.; Elms, S.C.; Clempus, R.E.; Campbell, D.J.; Sutliff, R.L.; Brown, L.A.; Guidot, D.M.; Hart, C.M. Chronic ethanol ingestion increases superoxide production and NADPH oxidase expression in the lung. Am. J. Respir. Cell Mol. Biol. 2006, 34, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Harris, F.L.; Hart, C.M.; Brown, L.A. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J. Immunol. 2012, 188, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.H.; Gallin, J.I.; Holland, S.M. The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 1995, 182, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, A.; Azoulay, E.; Marullo, S.; Flahault, A.; Fouqueray, B.; Philippe, C.; Cadranel, J.; Baud, L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am. J. Respir. Cell Mol. Biol. 1999, 21, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 2005, 111, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Nkabyo, Y.S.; Go, Y.M.; Ziegler, T.R.; Jones, D.P. Extracellular cysteine/cystine redox regulates the p44/p42 MAPK pathway by metalloproteinase-dependent epidermal growth factor receptor signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G70–G78. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Ramadan, B.; Ritzenthaler, J.D.; Rivera, H.N.; Jones, D.P.; Roman, J. Extracellular cysteine/cystine redox potential controls lung fibroblast proliferation and matrix expression through upregulation of transforming growth factor-beta. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L972–L981. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Ramirez, A.M.; Ritzenthaler, J.D.; Torres-Gonzalez, E.; Roser-Page, S.; Mora, A.L.; Brigham, K.L.; Jones, D.P.; Roman, J.; Rojas, M. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L37–L45. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Accardi, C.J.; Ziegler, T.R.; Blanco, R.A.; Ritzenthaler, J.D.; Rojas, M.; Roman, J.; Jones, D.P. Cysteine redox potential determines pro-inflammatory IL-1beta levels. PLoS ONE 2009, 4, e5017. [Google Scholar] [CrossRef]
- Clark, R.A.; Lanigan, J.M.; DellaPelle, P.; Manseau, E.; Dvorak, H.F.; Colvin, R.B. Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. J. Investig. Dermatol. 1982, 79, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, P.; Rennard, S.; Adelberg, S.; Crystal, R.G. Role of fibronectin in fibrotic lung disease. A growth factor for human lung fibroblasts. Chest 1983, 83, 96S. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.A.; Clark, R.A.F.; Swigart, L.M.; Huff, J.C.; Weston, W.M.; Howell, S.E. Fibronectin fragment(s) are chemotactic for human peripheral blood monocytes. J. Immunol. 1982, 129, 1612–1618. [Google Scholar]
- Burnham, E.L.; Moss, M.; Ritzenthaler, J.D.; Roman, J. Increased fibronectin expression in lung in the setting of chronic alcohol abuse. Alcohol. Clin. Exp. Res. 2007, 31, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Ritzenthaler, J.D.; Guidot, D.M.; Roman, J. Alveolar type II cells from ethanol-fed rats produce a fibronectin-enriched extracellular matrix that promotes monocyte activation. Alcohol 2007, 41, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Ward, C.; Findley, M.K.; Roser-Page, S.; Helms, M.N.; Roman, J. Extracellular matrix influences alveolar epithelial claudin expression and barrier function. Am. J. Respir. Cell Mol. Biol. 2010, 42, 172–180. [Google Scholar] [CrossRef]
- Pavlov, M. The anti-toxic function of the liver. Lancet 1893, 2, 1092. [Google Scholar]
- Rutenburg, A.M.; Sonnenblick, E.; Koven, I.; Aprahamian, H.A.; Reiner, L.; Fine, J. The role of intestinal bacteria in the development of dietary cirrhosis in rats. J. Exp. Med. 1957, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Broitman, S.A.; Gottlieb, L.S.; Zamcheck, N. Influence of neomycin and ingested endotoxin in the pathogenesis of choline deficiency cirrhosis in the adult rat. J. Exp. Med. 1964, 119, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P.; Ali, M.V. Endotoxin and the liver. II Effect of tolerance on carbon tetrachloride induced injury. J. Med. 1973, 4, 28–38. [Google Scholar] [PubMed]
- Nolan, J.P.; Leibowitz, A.I. Endotoxin and the liver. III. Modification of acute carbon tetrachloride injury by polymyxin B—An antiendotoxin. Gastroenterology 1978, 75, 445–449. [Google Scholar]
- Nolan, J.P. The role of intestinal endotoxin in liver injury: A long and evolving history. Hepatology 2010, 52, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.P.; Arroyo, V.; Gazzard, B.G.; Moodie, H.; Williams, R. Relation of renal impairment and haemorrhagic diathesis to endotoxaemia in fulminant hepatic failure. Lancet 1974, 1, 521–524. [Google Scholar] [CrossRef]
- Tarao, K.; Moroi, T.; Nagakura, Y.; Ikeuchi, T.; Suyama, T.; Endo, O.; Fukushima, K. Relationship between endotoxaemia and protein concentration of ascites in cirrhotic patients. Gut 1979, 20, 205–210. [Google Scholar] [CrossRef]
- Bode, C.H.; Kugler, V.; Bode, J.C.H. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Yin, M.; Bradford, B.U.; Wheeler, M.D.; Uesugi, T.; Froh, M.; Goyert, S.M.; Thurman, R.G. Reduced early alcohol-induced liver injury in CD14-deficient mice. J. Immunol. 2001, 166, 4737–4742. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol- induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Wheeler, M.D.; Gabele, E.; Isayama, F.; Thurman, R.G. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J. Immunol. 2002, 168, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Chen, W.C.; Schnabl, B. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front. Physiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; McClain, C.J. Probiotics in the treatment of the liver diseases. J. Am. Coll Nutr. 2012, 31, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Khanlou, H.; Souto, H.; Lippmann, M.; Munoz, S.; Rothstein, K.; Ozden, Z. Resolution of adult respiratory distress syndrome after recovery from fulminant hepatic failure. Am. J. Med. Sci. 1999, 317, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Matuschak, G.M. Lung-liver interactions in sepsis and multiple organ failure syndrome. Clin. Chest. Med. 1996, 17, 83–98. [Google Scholar] [CrossRef]
- Matuschak, G.M.; Pinsky, M.R.; Klein, E.C.; van Thiel, D.H.; Rinaldo, J.E. Effects of d-galactosamine-induced acute liver injury on mortality and pulmonary responses to Escherichia coli lipopolysaccharide. Modulation by arachidonic acid metabolites. Am. Rev. Respirat. Dis. 1990, 141, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Siore, A.M.; Parker, R.E.; Stecenko, A.A.; Cuppels, C.; McKean, M.; Christman, B.W.; Cruz-Gervis, R.; Brigham, K.L. Endotoxin-induced acute lung injury requires interaction with the liver. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L769–L776. [Google Scholar] [CrossRef] [PubMed]
- Siore, A.M.; Parker, R.E.; Cuppels, C.; Thorn, N.; Hansen, J.M.; Stecenko, A.A.; Brigham, K.L. The role of mitochondrial oxidation in endotoxin-induced liver-dependent swine pulmonary edema. Pulm. Pharmacol. Ther. 2012, 25, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Matuschak, G.M.; Mattingly, M.E.; Tredway, T.L.; Lechner, A.J. Liver-lung interactions during E. coli endotoxemia. TNF-alpha:leukotriene axis. Am. J. Respir. Crit Care Med. 1994, 149, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Massey, V.L.; Poole, L.G.; Siow, D.L.; Torres, E.; Warner, N.L.; Schmidt, R.H.; Ritzenthaler, J.D.; Roman, J.; Arteel, G.E. Chronic alcohol exposure enhances lipopolysaccharide-induced lung injury in mice: Potential role of systemic tumor necrosis factor alpha. Alcohol. Clin. Exp. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massey, V.L.; Beier, J.I.; Ritzenthaler, J.D.; Roman, J.; Arteel, G.E. Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology. Biomolecules 2015, 5, 2477-2503. https://doi.org/10.3390/biom5042477
Massey VL, Beier JI, Ritzenthaler JD, Roman J, Arteel GE. Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology. Biomolecules. 2015; 5(4):2477-2503. https://doi.org/10.3390/biom5042477
Chicago/Turabian StyleMassey, Veronica L., Juliane I. Beier, Jeffrey D. Ritzenthaler, Jesse Roman, and Gavin E. Arteel. 2015. "Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology" Biomolecules 5, no. 4: 2477-2503. https://doi.org/10.3390/biom5042477
APA StyleMassey, V. L., Beier, J. I., Ritzenthaler, J. D., Roman, J., & Arteel, G. E. (2015). Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology. Biomolecules, 5(4), 2477-2503. https://doi.org/10.3390/biom5042477