
The Molecular Circadian Clock and Alcohol-Induced Liver Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
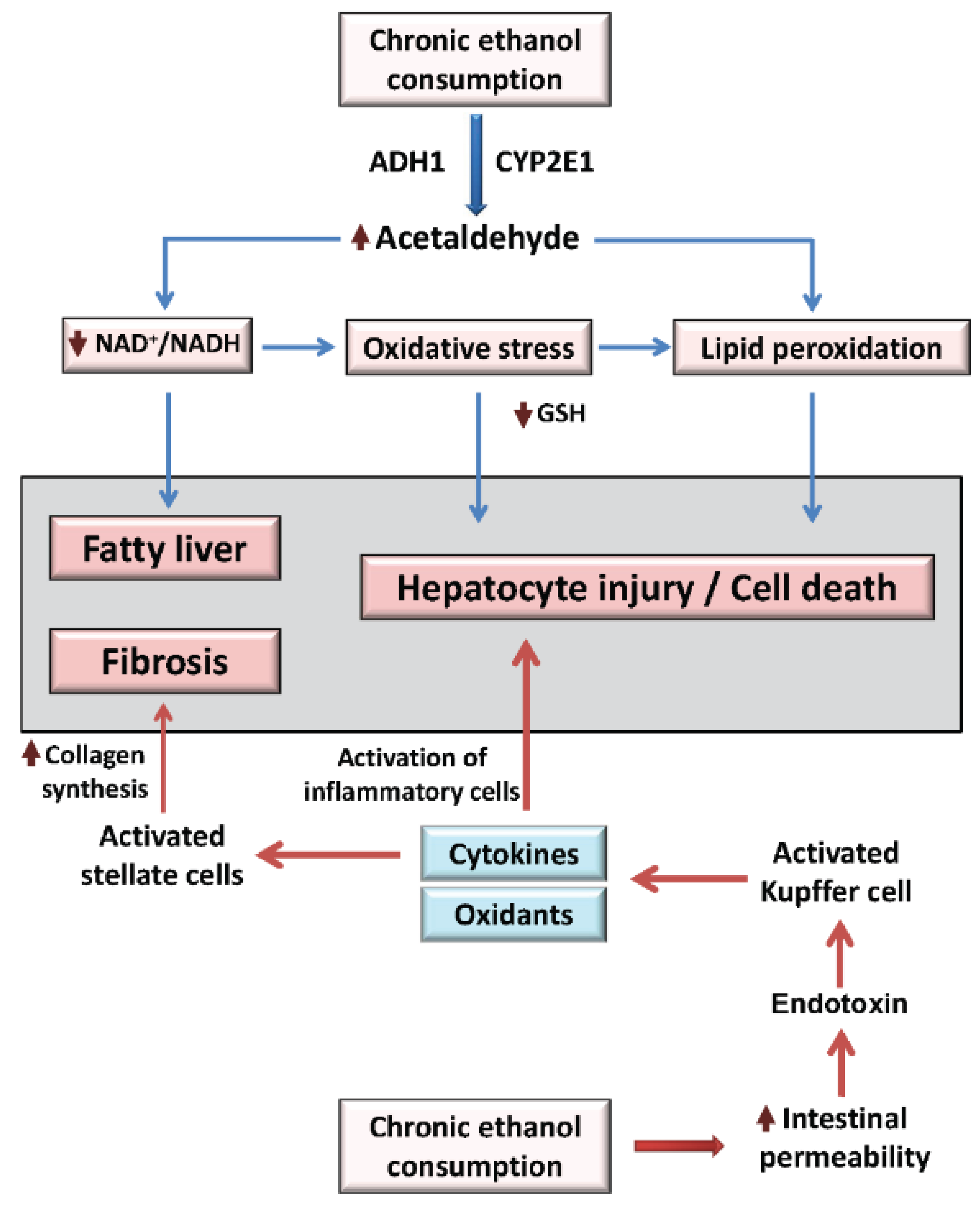
2. Overview of Alcoholic Liver Disease Mechanisms

3. The Molecular Circadian Clock and Metabolism
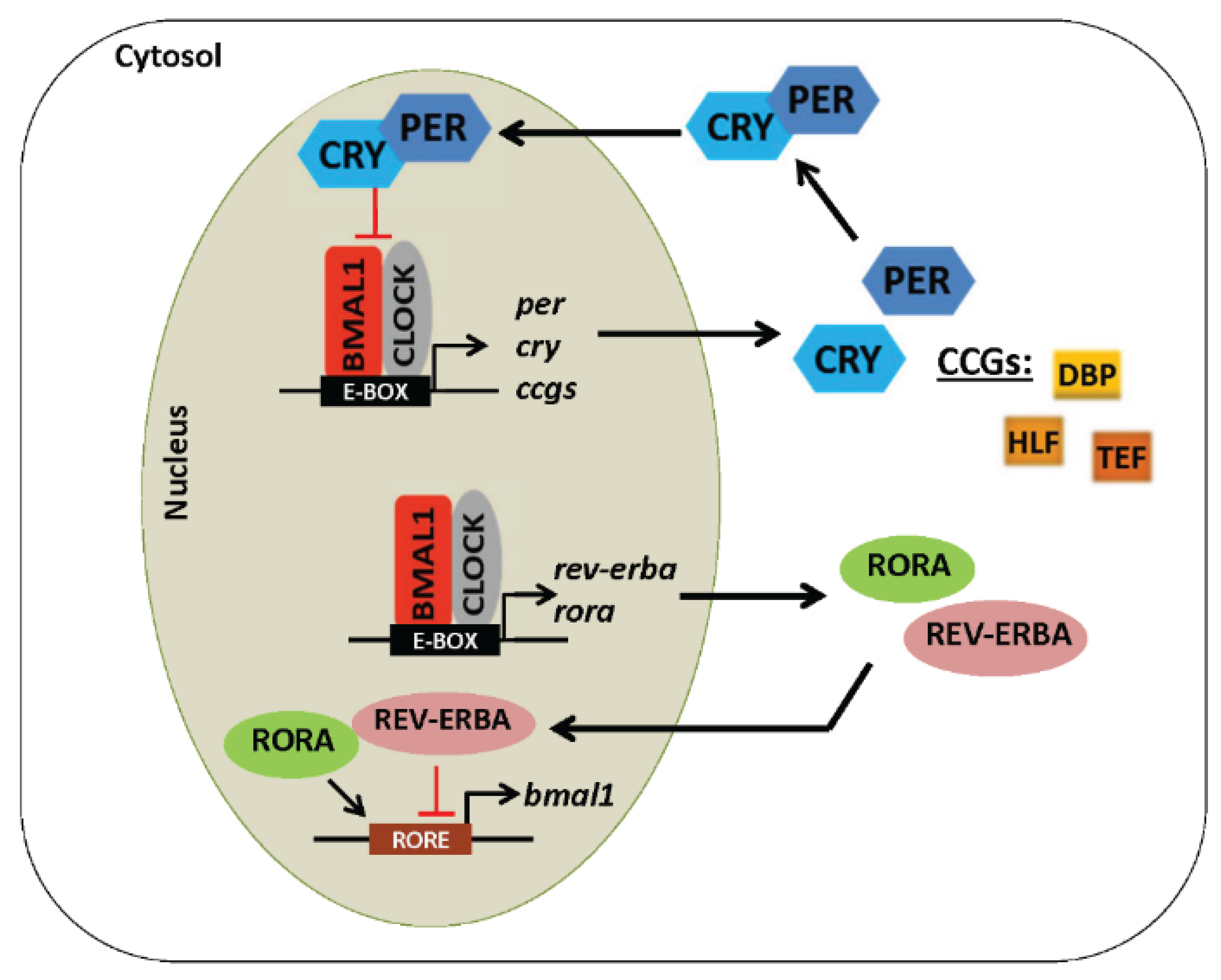
4. Core Components of the Molecular Circadian Clock

5. Regulation of the Circadian Clock
5.1. Cellular Redox Status and the Clock
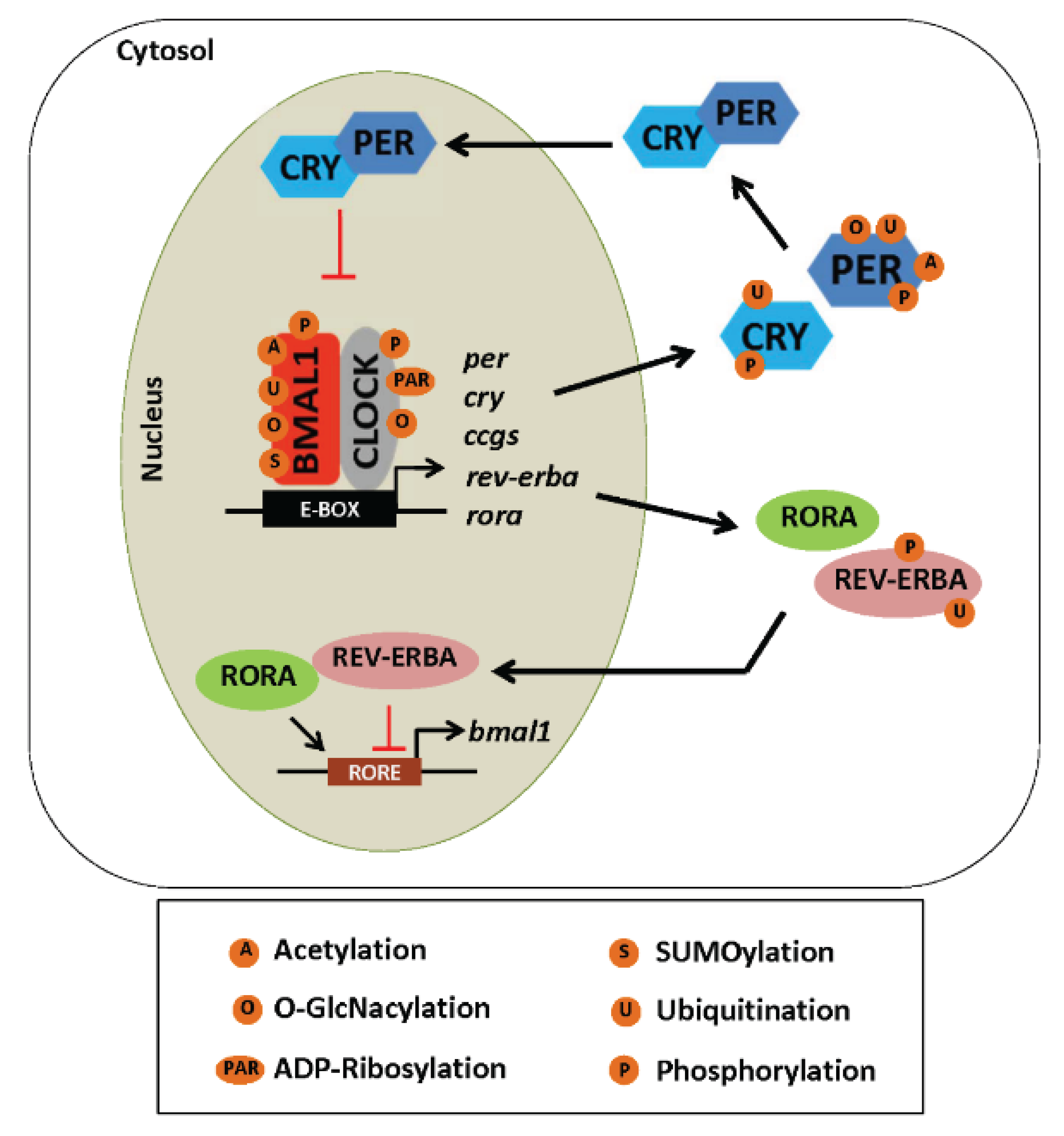
5.2. Post-Translational Modifications and the Clock

5.2.1. Phosphorylation
5.2.2. Acetylation
5.2.3. ADP-Ribosylation
5.2.4. Ubiquitination
5.2.5. SUMOylation
5.2.6. O-GlcNAcylation
5.3. Post-Transcriptional Mechanisms and the Clock
MicroRNAs and Alcoholic Liver Disease
6. Alcohol and Circadian Clocks
6.1. Role of Circadian Clocks in Alcohol-Induced Liver Injury
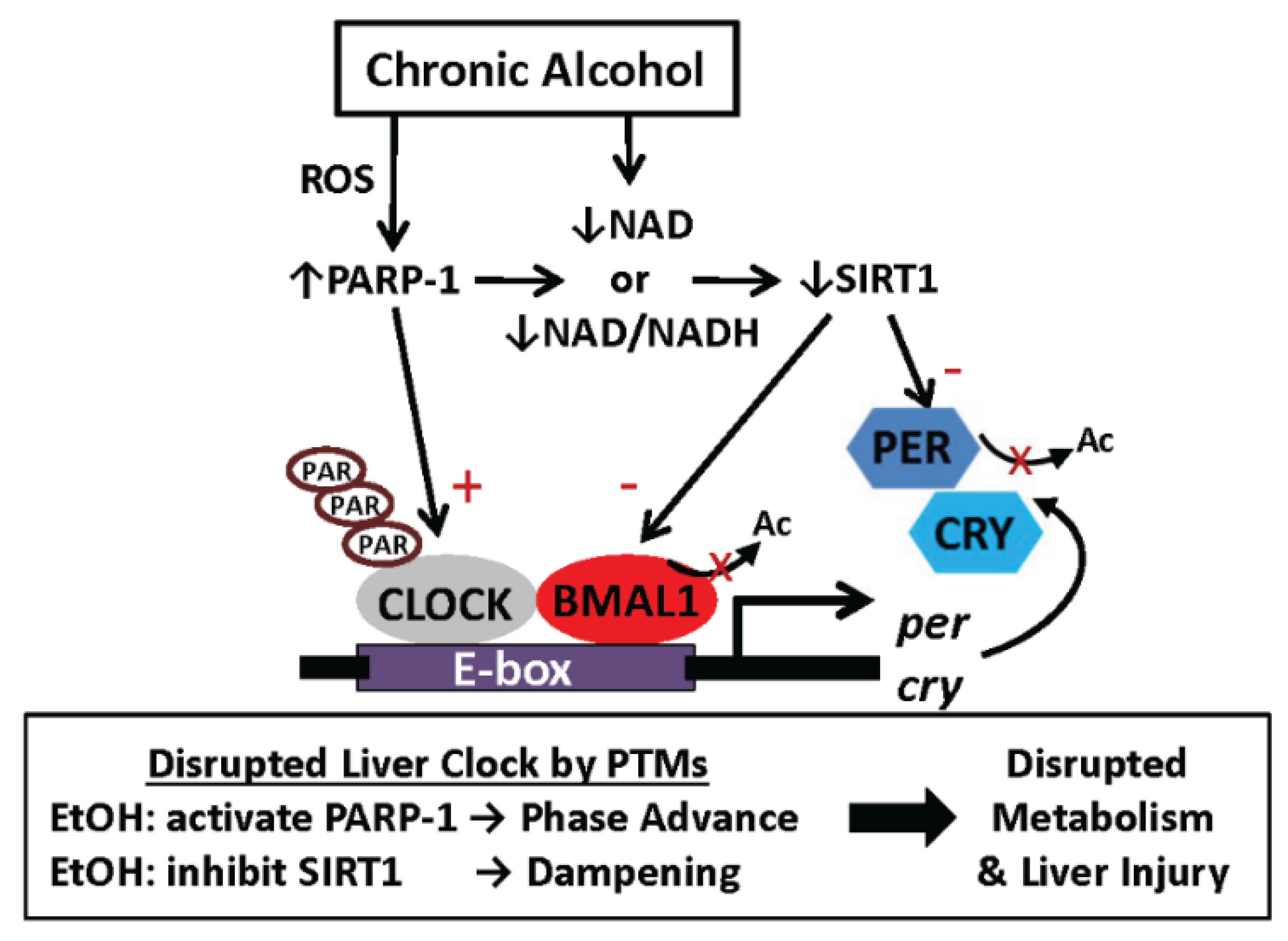
6.2. One Proposed Model for How Alcohol Disrupts Function of the Liver Clock: Focus on Protein Post-Translational Modifications

6.3. Other Possible Mechanisms Responsible for Alcohol-Mediated Alteration in Liver Clock Function
6.4. Potential Clock-Targeted Treatments for Alcoholic Liver Disease
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Danaei, G.; Ding, E.L.; Mozaffarian, D.; Taylor, B.; Rehm, J.; Murray, C.J.; Ezzati, M. The preventable causes of death in the united states: Comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 2009, 6, e1000058. [Google Scholar] [PubMed]
- Center for Disease Control and Prevention (CDC). Alcohol-Related Disease Impact (ARDI); CDC: Atlanta, GA, USA, 1989. [Google Scholar]
- Stahre, M.; Roeber, J.; Kanny, D.; Brewer, R.D.; Zhang, X. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev. Chronic Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bouchery, E.E.; Harwood, H.J.; Sacks, J.J.; Simon, C.J.; Brewer, R.D. Economic costs of excessive alcohol consumption in the U.S., 2006. Am. J. Prev. Med. 2011, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Substance Abuse and Mental Health Services Administration. Results from the 2003 National Survey on Drug Use and Health: National Findings; DHHS Publication No. SMA 04–3964; Office of Applied Studies: Rockville, MD, USA, 2004.
- Hasin, D.S.; Stinson, F.S.; Ogburn, E.; Grant, B.F. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: Results from the national epidemiologic survey on alcohol and related conditions. Arch. Gen. Psychiatry 2007, 64, 830–842. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Alcohol Abuse and Alcoholism. NIAAA Council Approves Definition of Binge Drinking. Available online: http://pubs.niaaa.nih.gov/publications/Newsletter/winter2004/Newsletter_Number3.pdf (accessed on 2 March 2015).
- Minino, A.M.; Heron, M.P.; Smith, B.L. Deaths: Preliminary data for 2004. Natl. Vital Stat. Rep. 2006, 54, 1–49. [Google Scholar] [PubMed]
- Day, C.P. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006, 26, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.N.; Millender-Swain, T.; Johnson, R., Jr.; Young, M.E.; Gamble, K.L.; Bailey, S.M. Chronic ethanol consumption disrupts the core molecular clock and diurnal rhythms of metabolic genes in the liver without affecting the suprachiasmatic nucleus. PLoS ONE 2013, 8, e71684. [Google Scholar] [CrossRef] [PubMed]
- Summa, K.C.; Voigt, R.M.; Forsyth, C.B.; Shaikh, M.; Cavanaugh, K.; Tang, Y.; Vitaterna, M.H.; Song, S.; Turek, F.W.; Keshavarzian, A. Disruption of the circadian clock in mice increases intestinal permeability and promotes alcohol-induced hepatic pathology and inflammation. PLoS ONE 2013, 8, e67102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ross, R.A.; Pywell, C.M.; Liangpunsakul, S.; Duffield, G.E. Disturbances in the murine hepatic circadian clock in alcohol-induced hepatic steatosis. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Werner, J.H.; Lee, D.; Sheppard, A.D.; Liangpunsakul, S.; Duffield, G.E. Dissociation between diurnal cycles in locomotor activity, feeding behavior and hepatic period2 expression in chronic alcohol-fed mice. Alcohol 2015, 49, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Udoh, U.S.; Swain, T.M.; Filiano, A.N.; Gamble, K.L.; Young, M.E.; Bailey, S.M. Chronic ethanol consumption disrupts diurnal rhythms of hepatic glycogen metabolism in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G964–G974. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Nagy, L.E. Pathogenesis of alcoholic liver disease: Interactions between parenchymal and non-parenchymal cells. J. Dig. Dis. 2011, 12, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Voigt, R.M.; Burgess, H.J.; Swanson, G.R.; Keshavarzian, A. Circadian rhythms, alcohol and gut interactions. Alcohol 2015, 49, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol Res. Curr. Rev. 2013, 35, 6–16. [Google Scholar]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.Q.; Ajmo, J.M.; You, M. Adiponectin and alcoholic fatty liver disease. IUBMB Life 2008, 60, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Shirai, H.; Ishida, N. Clock is involved in the circadian transactivation of peroxisome-proliferator-activated receptor alpha (pparalpha) in mice. Biochem. J. 2005, 386, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, Y.K.; Esterly, N.; Klaassen, C.D.; Wan, Y.J. Gender disparity of hepatic lipid homoeostasis regulated by the circadian clock. J. Biochem. 2009, 145, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates clock-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Cretenet, G.; Le Clech, M.; Gachon, F. Circadian clock-coordinated 12 h period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 2010, 11, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.B.; Karp, N.A.; Maywood, E.S.; Sage, E.A.; Deery, M.; O’Neill, J.S.; Wong, G.K.; Chesham, J.; Odell, M.; Lilley, K.S.; et al. Circadian orchestration of the hepatic proteome. Curr. Biol. 2006, 16, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L.; Patel, V.R.; Mohney, R.P.; Vignola, K.S.; Baldi, P.; Sassone-Corsi, P. Coordination of the transcriptome and metabolome by the circadian clock. Proc. Natl. Acad. Sci. USA 2012, 109, 5541–5546. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef]
- Bailey, S.M.; Udoh, U.S.; Young, M.E. Circadian regulation of metabolism. J. Endocrinol. 2014, 222, R75–R96. [Google Scholar] [CrossRef] [PubMed]
- Bass, J. Circadian topology of metabolism. Nature 2012, 491, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.A.; Weaver, D.R. Disrupting the circadian clock: Gene-specific effects on aging, cancer, and other phenotypes. Aging 2011, 3, 479–493. [Google Scholar] [PubMed]
- Lund, J.; Arendt, J.; Hampton, S.M.; English, J.; Morgan, L.M. Postprandial hormone and metabolic responses amongst shift workers in antarctica. J. Endocrinol. 2001, 171, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J.; Yang, J.N.; Scheer, F.A. The impact of the circadian timing system on cardiovascular and metabolic function. Prog. Brain Res. 2012, 199, 337–358. [Google Scholar] [PubMed]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Akashi, M.; Takumi, T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat. Struct. Mol. Biol. 2005, 12, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Preitner, N.; Damiola, F.; Lopez-Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, R.D.; McNamara, P.; Naik, K.A.; FitzGerald, G.A.; Kay, S.A.; Hogenesch, J.B. A functional genomics strategy reveals RORA as a component of the mammalian circadian clock. Neuron 2004, 43, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Bellet, M.M.; Sassone-Corsi, P. Mammalian circadian clock and metabolism—The epigenetic link. J. Cell Sci. 2010, 123, 3837–3848. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Fukada, Y. Resetting mechanism of central and peripheral circadian clocks in mammals. Zoolog. Sci. 2004, 21, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Stokkan, K.A.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the circadian clock in the liver by feeding. Science 2001, 291, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Damiola, F.; le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Arble, D.M.; Bass, J.; Laposky, A.D.; Vitaterna, M.H.; Turek, F.W. Circadian timing of food intake contributes to weight gain. Obesity 2009, 17, 2100–2102. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.S.; Ratcliffe, W.F.; Grenett, M.H.; Brewer, R.A.; Gamble, K.L.; Young, M.E. Quantitative analysis of light-phase restricted feeding reveals metabolic dyssynchrony in mice. Int. J. Obes. 2013, 37, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Duncan, M.J.; Esser, K.A. Chronic phase advance alters circadian physiological rhythms and peripheral molecular clocks. J. Appl. Physiol. 2013, 115, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in Bmal1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Schaad, O.; Reinke, H.; Saini, C.; Schibler, U. Regulation of circadian gene expression in liver by systemic signals and hepatocyte oscillators. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. Minireview: NAD+, a circadian metabolite with an epigenetic twist. Endocrinology 2012, 153, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.S.; Tsai, J.Y.; Villegas-Montoya, C.; Boland, B.B.; Blasier, Z.; Egbejimi, O.; Kueht, M.; Young, M.E. Time-of-day-dependent dietary fat consumption influences multiple cardiometabolic syndrome parameters in mice. Int. J. Obes. 2010, 34, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J.; Yang, J.N.; Garcia, J.I.; Myers, S.; Bozzi, I.; Wang, W.; Buxton, O.M.; Shea, S.A.; Scheer, F.A. Endogenous circadian system and circadian misalignment impact glucose tolerance via separate mechanisms in humans. Proc. Natl. Acad. Sci. USA 2015, 112, E2225–E2234. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Reick, M.; Wu, L.C.; McKnight, S.L. Regulation of Clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 2001, 293, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by Clock-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Cunningham, C.C. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 1998, 28, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.; Loudon, A.S.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef]
- Gallego, M.; Virshup, D.M. Post-translational modifications regulate the ticking of the circadian clock. Nat. Rev. Mol. Cell Biol. 2007, 8, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.J.; Logunova, L.; Maywood, E.S.; Gallego, M.; Lebiecki, J.; Brown, T.M.; Sladek, M.; Semikhodskii, A.S.; Glossop, N.R.; Piggins, H.D.; et al. Setting clock speed in mammals: The CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing period proteins. Neuron 2008, 58, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.C.; Vanselow, J.T.; Kramer, A.; Edery, I. The phospho-occupancy of an atypical slimb-binding site on period that is phosphorylated by doubletime controls the pace of the clock. Genes Dev. 2008, 22, 1758–1772. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Machida, K.K.; Noton, E.; Constance, C.M.; Dallmann, R.; di Napoli, M.N.; DeBruyne, J.P.; Lambert, C.M.; Yu, E.A.; Reppert, S.M.; et al. Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol. Cell. Biol. 2009, 29, 3853–3866. [Google Scholar] [CrossRef] [PubMed]
- Eide, E.J.; Woolf, M.F.; Kang, H.; Woolf, P.; Hurst, W.; Camacho, F.; Vielhaber, E.L.; Giovanni, A.; Virshup, D.M. Control of mammalian circadian rhythm by CK1epsilon-regulated proteasome-mediated PER2 degradation. Mol. Cell. Biol. 2005, 25, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (PER1) protein. J. Biol. Chem. 2005, 280, 26863–26872. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, E.; Eide, E.; Rivers, A.; Gao, Z.H.; Virshup, D.M. Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon. Mol. Cell. Biol. 2000, 20, 4888–4899. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Bassermann, F.; Maiolica, A.; Lee, C.; Nolan, P.M.; Godinho, S.I.; Draetta, G.F.; Pagano, M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science 2007, 316, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.I.; Maywood, E.S.; Shaw, L.; Tucci, V.; Barnard, A.R.; Busino, L.; Pagano, M.; Kendall, R.; Quwailid, M.M.; Romero, M.R.; et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 2007, 316, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Siepka, S.M.; Yoo, S.H.; Park, J.; Song, W.; Kumar, V.; Hu, Y.; Lee, C.; Takahashi, J.S. Circadian mutant overtime reveals f-box protein Fbxl3 regulation of cryptochrome and period gene expression. Cell 2007, 129, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Eide, E.J.; Vielhaber, E.L.; Hinz, W.A.; Virshup, D.M. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase 1 epsilon. J. Biol. Chem. 2002, 277, 17248–17254. [Google Scholar] [CrossRef] [PubMed]
- Sanada, K.; Okano, T.; Fukada, Y. Mitogen-activated protein kinase phosphorylates and negatively regulates basic helix-loop-helix-PAS transcription factor BMAL1. J. Biol. Chem. 2002, 277, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Besing, R.C.; Paul, J.R.; Hablitz, L.M.; Rogers, C.O.; Johnson, R.L.; Young, M.E.; Gamble, K.L. Circadian rhythmicity of active GSK3 isoforms modulates molecular clock gene rhythms in the suprachiasmatic nucleus. J. Biol. Rhythms 2015, 30, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Iitaka, C.; Miyazaki, K.; Akaike, T.; Ishida, N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J. Biol. Chem. 2005, 280, 29397–29402. [Google Scholar] [CrossRef] [PubMed]
- Durgan, D.J.; Young, M.E. The cardiomyocyte circadian clock: Emerging roles in health and disease. Circ. Res. 2010, 106, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Zocchi, L.; Kinoshita, C.; Borrelli, E.; Sassone-Corsi, P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE 2010, 5, e8561. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wang, J.; Klein, P.S.; Lazar, M.A. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science 2006, 311, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Sakai, M.; Kurabayashi, N.; Hirota, T.; Fukada, Y. Ser-557-phosphorylated mCry2 is degraded upon synergistic phosphorylation by glycogen synthase kinase-3 beta. J. Biol. Chem. 2005, 280, 31714–31721. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator clock is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. Clock-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Leo, M.A.; Wang, X.; Decarli, L.M. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC-1alpha in rats. Biochem. Biophys. Res. Commun. 2008, 370, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Zhang, R.; Shen, Z.; Flatow, L.; You, M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J. Biol. Chem. 2012, 287, 9817–9826. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of poly(ADP-ribose) polymerases: The factotums of cell maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Reinke, H.; Altmeyer, M.; Gutierrez-Arcelus, M.; Hottiger, M.O.; Schibler, U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 2010, 142, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Schibler, U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011, 13, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Yumimoto, K.; Tsunematsu, R.; Matsumoto, M.; Oyama, M.; Kozuka-Hata, H.; Nakagawa, T.; Lanjakornsiripan, D.; Nakayama, K.I.; Fukada, Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 2013, 152, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of Cry in nucleus and cytoplasm. Cell 2013, 152, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, Y.; Lee, M.J.; Park, E.; Kang, S.H.; Chung, C.H.; Lee, K.H.; Kim, K. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the Clock/BMAL1 complex. Mol. Cell. Biol. 2008, 28, 6056–6065. [Google Scholar] [CrossRef] [PubMed]
- Scoma, H.D.; Humby, M.; Yadav, G.; Zhang, Q.; Fogerty, J.; Besharse, J.C. The de-ubiquitinylating enzyme, USP2, is associated with the circadian clockwork and regulates its sensitivity to light. PLoS ONE 2011, 6, e25382. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Duguay, D.; Fahrenkrug, J.; Cermakian, N.; Wing, S.S. USP2 regulates the intracellular localization of PER1 and circadian gene expression. J. Biol. Rhythms 2014, 29, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Cardone, L.; Hirayama, J.; Giordano, F.; Tamaru, T.; Palvimo, J.J.; Sassone-Corsi, P. Circadian clock control by sumoylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, M.L.; Tomasi, I.; Ramani, K.; Pascale, R.M.; Xu, J.; Giordano, P.; Mato, J.M.; Lu, S.C. S-Adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology 2012, 56, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Jang, H.; Lee, J.H.; Huh, J.Y.; Choi, S.; Chung, J.; Kim, J.B. PIASy-mediated sumoylation of SREBP1C regulates hepatic lipid metabolism upon fasting signaling. Mol. Cell. Biol. 2014, 34, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Xiao, Z.; Kwon, S.; Sun, X.; Ryerson, D.; Tkac, D.; Ma, P.; Wu, S.Y.; Chiang, C.M.; Zhou, E.; et al. A dysregulated ACETYL/SUMO switch of Fxr promotes hepatic inflammation in obesity. EMBO J. 2015, 34, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Durgan, D.J.; Pat, B.M.; Laczy, B.; Bradley, J.A.; Tsai, J.Y.; Grenett, M.H.; Ratcliffe, W.F.; Brewer, R.A.; Nagendran, J.; Villegas-Montoya, C.; et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J. Biol. Chem. 2011, 286, 44606–44619. [Google Scholar] [CrossRef] [PubMed]
- Li, M.D.; Ruan, H.B.; Hughes, M.E.; Lee, J.S.; Singh, J.P.; Jones, S.P.; Nitabach, M.N.; Yang, X. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/Clock ubiquitination. Cell Metab. 2013, 17, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Yoo, S.H.; Huang, H.C.; Kumar, V.; Lee, C.; Kim, T.K.; Takahashi, J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, K.H.; Kim, D.Y.; Kwak, E.; Kim, S.; Kim, K.T. Rhythmic control of mRNA stability modulates circadian amplitude of mouse period3 mRNA. J. Neurochem. 2015, 132, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.C.; Ha, D.C.; Lee, K.H.; Kim, D.Y.; Kim, T.D.; Kim, K.T. Circadian amplitude of cryptochrome 1 is modulated by mRNA stability regulation via cytoplasmic hnRNP D oscillation. Mol. Cell. Biol. 2010, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Morf, J.; Rey, G.; Schneider, K.; Stratmann, M.; Fujita, J.; Naef, F.; Schibler, U. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science 2012, 338, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Baggs, J.E.; Green, C.B. Nocturnin, a deadenylase in xenopus laevis retina: A mechanism for posttranscriptional control of circadian-related mRNA. Curr. Biol. 2003, 13, 189–198. [Google Scholar] [CrossRef]
- Baggs, J.E.; Green, C.B. Functional analysis of nocturnin: A circadian clock-regulated gene identified by differential display. Methods Mol. Biol. 2006, 317, 243–254. [Google Scholar] [PubMed]
- Kojima, S.; Sher-Chen, E.L.; Green, C.B. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 2012, 26, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Douris, N.; Green, C.B. Noc out the fat: A short review of the circadian deadenylase nocturnin. Ann. Med. 2008, 40, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Gatfield, D.; Esau, C.C.; Green, C.B. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase nocturnin in mouse liver. PLoS ONE 2010, 5, e11264. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Shingle, D.L.; Green, C.B. Post-transcriptional control of circadian rhythms. J. Cell Sci. 2011, 124, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Cheng, H.Y. Micro-managing the circadian clock: The role of microRNAs in biological timekeeping. J. Mol. Biol. 2013, 425, 3609–3624. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. MicroRNA modulation of circadian-clock period and entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Shende, V.R.; Goldrick, M.M.; Ramani, S.; Earnest, D.J. Expression and rhythmic modulation of circulating microRNAs targeting the clock gene Bmal1 in mice. PLoS ONE 2011, 6, e22586. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Clijsters, L.; Agami, R. The miRNA-192/194 cluster regulates the period gene family and the circadian clock. FEBS J. 2009, 276, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; D’Alessandro, M.; Lee, C. MiRNAs are required for generating a time delay critical for the circadian oscillator. Curr. Biol. 2013, 23, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Gatfield, D.; le Martelot, G.; Vejnar, C.E.; Gerlach, D.; Schaad, O.; Fleury-Olela, F.; Ruskeepaa, A.L.; Oresic, M.; Esau, C.C.; Zdobnov, E.M.; et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev. 2009, 23, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Douris, N.; Kojima, S.; Pan, X.; Lerch-Gaggl, A.F.; Duong, S.Q.; Hussain, M.M.; Green, C.B. Nocturnin regulates circadian trafficking of dietary lipid in intestinal enterocytes. Curr. Biol. 2011, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Douris, N.; Kojima, S.; Strayer, C.A.; Fogerty, J.; Lourim, D.; Keller, S.R.; Besharse, J.C. Loss of nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 9888–9893. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, K.; Herrera, L.; Zhou, T.; Francis, H.; Han, Y.; Levine, P.; Lin, E.; Glaser, S.; Alpini, G.; Meng, F. The functional role of microRNAs in alcoholic liver injury. J. Cell. Mol. Med. 2014, 18, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Satishchandran, A. MicroRNAs in alcoholic liver disease. Semin. Liver Dis. 2015, 35, 36–42. [Google Scholar] [PubMed]
- Yin, H.; Liang, X.; Jogasuria, A.; Davidson, N.O.; You, M. Mir-217 regulates ethanol-induced hepatic inflammation by disrupting sirtuin 1-lipin-1 signaling. Am. J. Pathol. 2015, 185, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor alpha (TNF-alpha) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; la Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Lippai, D.; Bala, S.; Catalano, D.; Kodys, K.; Szabo, G. Micro-RNA-155 deficiency prevents alcohol-induced serum endotoxin increase and small bowel inflammation in mice. Alcohol Clin. Exp. Res. 2014, 38, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Lippai, D.; Bala, S.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS ONE 2013, 8, e70945. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.M.; Fagundes, C.T.; Yang, G.; Palsson-McDermott, E.M.; Wochal, P.; McGettrick, A.F.; Foley, N.H.; Early, J.O.; Chen, L.; Zhang, H.; et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. USA 2015, 112, 7231–7236. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Reddy, A.B. Rethinking the clockwork: Redox cycles and non-transcriptional control of circadian rhythms. Biochem. Soc. Trans. 2014, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosenwasser, A.M. Circadian clock genes: Non-circadian roles in sleep, addiction, and psychiatric disorders? Neurosci. Biobehav. Rev. 2010, 34, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Hasler, B.P.; Clark, D.B. Circadian misalignment, reward-related brain function, and adolescent alcohol involvement. Alcohol Clin. Exp. Res. 2013, 37, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Damaggio, A.S.; Gorman, M.R. The circadian timing system in ethanol consumption and dependence. Behav. Neurosci. 2014, 128, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Prosser, R.A.; Mangrum, C.A.; Glass, J.D. Acute ethanol modulates glutamatergic and serotonergic phase shifts of the mouse circadian clock in vitro. Neuroscience 2008, 152, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Logan, R.W.; Seggio, J.A.; Robinson, S.L.; Richard, G.R.; Rosenwasser, A.M. Circadian wheel-running activity during withdrawal from chronic intermittent ethanol exposure in mice. Alcohol 2010, 44, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Brager, A.J.; Ruby, C.L.; Prosser, R.A.; Glass, J.D. Chronic ethanol disrupts circadian photic entrainment and daily locomotor activity in the mouse. Alcohol Clin. Exp. Res. 2010, 34, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Brager, A.J.; Ruby, C.L.; Prosser, R.A.; Glass, J.D. Acute ethanol disrupts photic and serotonergic circadian clock phase-resetting in the mouse. Alcohol Clin. Exp. Res. 2011, 35, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Danel, T.; Jeanson, R.; Touitou, Y. Temporal pattern in consumption of the first drink of the day in alcohol-dependent persons. Chronobiol. Int. 2003, 20, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Schluter, P.J.; Turner, C.; Benefer, C. Long working hours and alcohol risk among australian and new zealand nurses and midwives: A cross-sectional study. Int. J. Nurs. Stud. 2012, 49, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Trinkoff, A.M.; Storr, C.L. Work schedule characteristics and substance use in nurses. Am. J. Ind. Med. 1998, 34, 266–271. [Google Scholar] [CrossRef]
- Virtanen, M.; Jokela, M.; Nyberg, S.T.; Madsen, I.E.; Lallukka, T.; Ahola, K.; Alfredsson, L.; Batty, G.D.; Bjorner, J.B.; Borritz, M.; et al. Long working hours and alcohol use: Systematic review and meta-analysis of published studies and unpublished individual participant data. Br. Med. J. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, Y.; Nakamura, K.; Sakurai, M.; Nagasawa, S.Y.; Ishizaki, M.; Nakashima, M.; Kido, T.; Naruse, Y.; Nakagawa, H. The effect of age on the relationships between work-related factors and heavy drinking. J. Occup. Health 2014, 56, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Gauvin, D.V.; Baird, T.J.; Vanecek, S.A.; Briscoe, R.J.; Vallett, M.; Holloway, F.A. Effects of time-of-day and photoperiod phase shifts on voluntary ethanol consumption in rats. Alcohol Clin. Exp. Res. 1997, 21, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.B.; Ruby, C.L.; Brager, A.J.; Prosser, R.A.; Glass, J.D. Environmental modulation of alcohol intake in hamsters: Effects of wheel running and constant light exposure. Alcohol Clin. Exp. Res. 2010, 34, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Rosenwasser, A.M.; Clark, J.W.; Fixaris, M.C.; Belanger, G.V.; Foster, J.A. Effects of repeated light-dark phase shifts on voluntary ethanol and water intake in male and female fischer and lewis rats. Alcohol 2010, 44, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Ozburn, A.R.; Gordon, E.A.; McClung, C.A. Clock delta 19 mutants exhibit increased ethanol preference and consumption. Alcohol Clin. Exp. Res. 2010, 34, 116A. [Google Scholar]
- Spanagel, R.; Pendyala, G.; Abarca, C.; Zghoul, T.; Sanchis-Segura, C.; Magnone, M.C.; Lascorz, J.; Depner, M.; Holzberg, D.; Soyka, M.; et al. The clock gene PER2 influences the glutamatergic system and modulates alcohol consumption. Nat. Med. 2005, 11, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Blomeyer, D.; Buchmann, A.F.; Lascorz, J.; Zimmermann, U.S.; Esser, G.; Desrivieres, S.; Schmidt, M.H.; Banaschewski, T.; Schumann, G.; Laucht, M. Association of PER2 genotype and stressful life events with alcohol drinking in young adults. PLoS ONE 2013, 8, e59136. [Google Scholar] [CrossRef] [PubMed]
- Comasco, E.; Nordquist, N.; Gokturk, C.; Aslund, C.; Hallman, J.; Oreland, L.; Nilsson, K.W. The clock gene PER2 and sleep problems: Association with alcohol consumption among Swedish adolescents. Ups. J. Med. Sci. 2010, 115, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gamble, K.L.; Motsinger-Reif, A.A.; Hida, A.; Borsetti, H.M.; Servick, S.V.; Ciarleglio, C.M.; Robbins, S.; Hicks, J.; Carver, K.; Hamilton, N.; et al. Shift work in nurses: Contribution of phenotypes and genotypes to adaptation. PLoS ONE 2011, 6, e18395. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, L.; Saarikoski, S.T.; Haukka, J.; Pirkola, S.; Aromaa, A.; Lonnqvist, J.; Partonen, T. Circadian clock gene polymorphisms in alcohol use disorders and alcohol consumption. Alcohol Alcohol. 2010, 45, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Perreau-Lenz, S.; Spanagel, R. Clock genes × stress × reward interactions in alcohol and substance use disorders. Alcohol 2015, 49, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mozhui, K.; Li, Z.; Mulligan, M.K.; Ingels, J.F.; Zhou, X.; Hori, R.T.; Chen, H.; Cook, M.N.; Williams, R.W.; et al. A promoter polymorphism in the Per3 gene is associated with alcohol and stress response. Transl. Psychiatry 2012. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Bechtold, D.A. The cost of circadian desynchrony: Evidence, insights and open questions. Bioessays 2015, 37, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. The feeding of alcohol in liquid diets: Two decades of applications and 1982 update. Alcohol. Clin. Exp. Res. 1982, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhang, D.; Arthurs, B.; Li, P.; Durudogan, L.; Gupta, N.; Yin, L. Palmitate inhibits SIRT1-dependent Bmal1/Clock interaction and disrupts circadian gene oscillations in hepatocytes. PLoS ONE 2015, 10, e0130047. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Yamazaki, S.; Lowrey, P.L.; Shimomura, K.; Ko, C.H.; Buhr, E.D.; Siepka, S.M.; Hong, H.K.; Oh, W.J.; Yoo, O.J.; et al. Period2::Luciferase real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. USA 2004, 101, 5339–5346. [Google Scholar] [CrossRef] [PubMed]
- Besing, R.C.; Hablitz, L.M.; Paul, J.R.; Johnson, R.L.; Prosser, R.A.; Gamble, K.L. Neuropeptide Y-induced phase shifts of Per2::Luc rhythms are mediated by long-term suppression of neuronal excitability in a phase-specific manner. Chronobiol. Int. 2012, 29, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Barnea, M.; Haviv, L.; Gutman, R.; Chapnik, N.; Madar, Z.; Froy, O. Metformin affects the circadian clock and metabolic rhythms in a tissue-specific manner. Biochim. Biophys. Acta 2012, 1822, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Tamagawa, T.; Shibata, S. Effect of chronic ethanol exposure on the liver of clock-mutant mice. J. Circadian Rhythms 2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yang, P.; Zhan, Y.; Xia, L.; Hua, Z.; Zhang, J. Deletion of circadian gene Per1 alleviates acute ethanol-induced hepatotoxicity in mice. Toxicology 2013, 314, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, C.G.; Cunningham, C.C. Contributions of dietary carbohydrate and ethanol to alterations in liver glycogen levels and glycolytic activity. Alcohol 1999, 19, 139–144. [Google Scholar] [CrossRef]
- Winston, G.W.; Reitz, R.C. Chronic ethanol ingestion and glycogen metabolism in male and female rats. Adv. Exp. Med. Biol. 1980, 132, 569–577. [Google Scholar] [PubMed]
- Farnell, Y.Z.; Allen, G.C.; Nahm, S.S.; Neuendorff, N.; West, J.R.; Chen, W.J.; Earnest, D.J. Neonatal alcohol exposure differentially alters clock gene oscillations within the suprachiasmatic nucleus, cerebellum, and liver of adult rats. Alcohol Clin. Exp. Res. 2008, 32, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Kang, P.; Faske, J.; Gomez-Acevedo, H.; Blackburn, M.L.; Badger, T.M.; Shankar, K. High fat diet and in utero exposure to maternal obesity disrupts circadian rhythm and leads to metabolic programming of liver in rat offspring. PLoS ONE 2014, 9, e84209. [Google Scholar] [PubMed]
- Swanson, G.; Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Turek, F.W.; Keshavarzian, A. Role of intestinal circadian genes in alcohol-induced gut leakiness. Alcohol Clin. Exp. Res. 2011, 35, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Schibler, U. Circadian timing of metabolism in animal models and humans. J. Intern. Med. 2015, 277, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.D.; Lamia, K.A. AMPK at the crossroads of circadian clocks and metabolism. Mol. Cell Endocrinol. 2013, 366, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes. Metab. 2011, 13, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Lo Sasso, G.; Moschetta, A.; Schibler, U. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar] [CrossRef] [PubMed]
- Akinshola, B.E.; Sharma, S.; Potter, J.J.; Mezey, E. Ethanol enhances ADP-ribosylation of protein in rat hepatocytes. Hepatology 1992, 15, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Yaguchi, M.; Itoga And, S.; Noda, M. Effects of chronic alcohol consumption on hepatic poly-ADP-ribosylation in the rat. Alcohol Clin. Exp. Res. 2001, 25, S35–S38. [Google Scholar] [CrossRef]
- Yang, S.; Koteish, A.; Lin, H.; Huang, J.; Roskams, T.; Dawson, V.; Diehl, A.M. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology 2004, 39, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. Interference between PARPs and SIRT1: A novel approach to healthy ageing? Aging 2011, 3, 543–547. [Google Scholar] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Green, M.F.; Petersen, D.R.; Hirschey, M.D. Ethanol metabolism modifies hepatic protein acylation in mice. PLoS ONE 2013, 8, e75868. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; Landar, A.; Davis, A.J.; Chamlee, L.; Sanderson, T.; Kim, H.; Page, G.; Pompilius, M.; Ballinger, S.; Darley-Usmar, V.; et al. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem. 2004, 279, 22092–22101. [Google Scholar] [CrossRef] [PubMed]
- Andringa, K.K.; King, A.L.; Eccleston, H.B.; Mantena, S.K.; Landar, A.; Jhala, N.C.; Dickinson, D.A.; Squadrito, G.L.; Bailey, S.M. Analysis of the liver mitochondrial proteome in response to ethanol and S-adenosylmethionine treatments: Novel molecular targets of disease and hepatoprotection. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G732–G745. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Cunningham, C.C. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcohol Clin. Exp. Res. 1999, 23, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Ivester, P.; Lide, M.J.; Cunningham, C.C. Effect of chronic ethanol consumption on the energy state and structural stability of periportal and perivenous hepatocytes. Arch. Biochem. Biophys. 1995, 322, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Um, J.H.; Yang, S.; Yamazaki, S.; Kang, H.; Viollet, B.; Foretz, M.; Chung, J.H. Activation of 5'-AMP-activated kinase with diabetes drug metformin induces casein kinase 1epsilon (CK1epsilon)-dependent degradation of clock protein mPer2. J. Biol. Chem. 2007, 282, 20794–20798. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Kawaratani, H.; Tsujimoto, T.; Douhara, A.; Takaya, H.; Moriya, K.; Namisaki, T.; Noguchi, R.; Yoshiji, H.; Fujimoto, M.; Fukui, H. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J.; Bala, S. Innate immunity and alcoholic liver disease. Dig. Dis. 2012, 30, S55–S60. [Google Scholar] [CrossRef] [PubMed]
- Petrzilka, S.; Taraborrelli, C.; Cavadini, G.; Fontana, A.; Birchler, T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J. Biol. Rhythms 2009, 24, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, G.; Petrzilka, S.; Kohler, P.; Jud, C.; Tobler, I.; Birchler, T.; Fontana, A. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Straub, R.H. Disruption of rhythms of molecular clocks in primary synovial fibroblasts of patients with osteoarthritis and rheumatoid arthritis, role of IL-1beta/TNF. Arthritis Res. Ther. 2012, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.; Meier, D.; Muller, A.; Franken, P.; Fujita, J.; Fontana, A. Tumor necrosis factor and transforming growth factor beta regulate clock genes by controlling the expression of the cold inducible RNA-binding protein (CIRBP). J. Biol. Chem. 2014, 289, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Paladino, N.; Mul Fedele, M.L.; Duhart, J.M.; Marpegan, L.; Golombek, D.A. Modulation of mammalian circadian rhythms by tumor necrosis factor-alpha. Chronobiol. Int. 2014, 31, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H.; Higashitsuji, H.; Yokoi, H.; Itoh, K.; Danno, S.; Matsuda, T.; Fujita, J. Cloning and characterization of human CIRP (cold-inducible rna-binding protein) cDNA and chromosomal assignment of the gene. Gene 1997, 204, 115–120. [Google Scholar] [CrossRef]
- Nishiyama, H.; Itoh, K.; Kaneko, Y.; Kishishita, M.; Yoshida, O.; Fujita, J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J. Cell Biol. 1997, 137, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Paladino, N.; Leone, M.J.; Plano, S.A.; Golombek, D.A. Paying the circadian toll: The circadian response to LPS injection is dependent on the toll-like receptor 4. J. Neuroimmunol. 2010, 225, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Rajayer, S.R.; Jacob, A.; Yang, W.L.; Zhou, M.; Chaung, W.; Wang, P. Cold-inducible RNA-binding protein is an important mediator of alcohol-induced brain inflammation. PLoS ONE 2013, 8, e79430. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.J.; Maywood, E.S.; Bechtold, D.A.; Lu, W.Q.; Li, J.; Gibbs, J.E.; Dupre, S.M.; Chesham, J.E.; Rajamohan, F.; Knafels, J.; et al. Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 15240–15245. [Google Scholar] [CrossRef] [PubMed]
- Perreau-Lenz, S.; Vengeliene, V.; Noori, H.R.; Merlo-Pich, E.V.; Corsi, M.A.; Corti, C.; Spanagel, R. Inhibition of the casein-kinase-1-epsilon/delta/ prevents relapse-like alcohol drinking. Neuropsychopharmacol 2012, 37, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Innominato, P.F.; Roche, V.P.; Palesh, O.G.; Ulusakarya, A.; Spiegel, D.; Levi, F.A. The circadian timing system in clinical oncology. Ann. Med. 2014, 46, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.K.; Chao, Y.C.; Tang, H.S.; Lang, H.F.; Hsu, C.T. Effects of extra-carbohydrate supplementation in the late evening on energy expenditure and substrate oxidation in patients with liver cirrhosis. J. Parenter. Enteral Nutr. 1997, 21, 96–99. [Google Scholar] [CrossRef]
- Yamanaka-Okumura, H.; Nakamura, T.; Takeuchi, H.; Miyake, H.; Katayama, T.; Arai, H.; Taketani, Y.; Fujii, M.; Shimada, M.; Takeda, E. Effect of late evening snack with rice ball on energy metabolism in liver cirrhosis. Eur. J. Clin. Nutr. 2006, 60, 1067–1072. [Google Scholar] [CrossRef]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udoh, U.S.; Valcin, J.A.; Gamble, K.L.; Bailey, S.M. The Molecular Circadian Clock and Alcohol-Induced Liver Injury. Biomolecules 2015, 5, 2504-2537. https://doi.org/10.3390/biom5042504
Udoh US, Valcin JA, Gamble KL, Bailey SM. The Molecular Circadian Clock and Alcohol-Induced Liver Injury. Biomolecules. 2015; 5(4):2504-2537. https://doi.org/10.3390/biom5042504
Chicago/Turabian StyleUdoh, Uduak S., Jennifer A. Valcin, Karen L. Gamble, and Shannon M. Bailey. 2015. "The Molecular Circadian Clock and Alcohol-Induced Liver Injury" Biomolecules 5, no. 4: 2504-2537. https://doi.org/10.3390/biom5042504
APA StyleUdoh, U. S., Valcin, J. A., Gamble, K. L., & Bailey, S. M. (2015). The Molecular Circadian Clock and Alcohol-Induced Liver Injury. Biomolecules, 5(4), 2504-2537. https://doi.org/10.3390/biom5042504