
Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
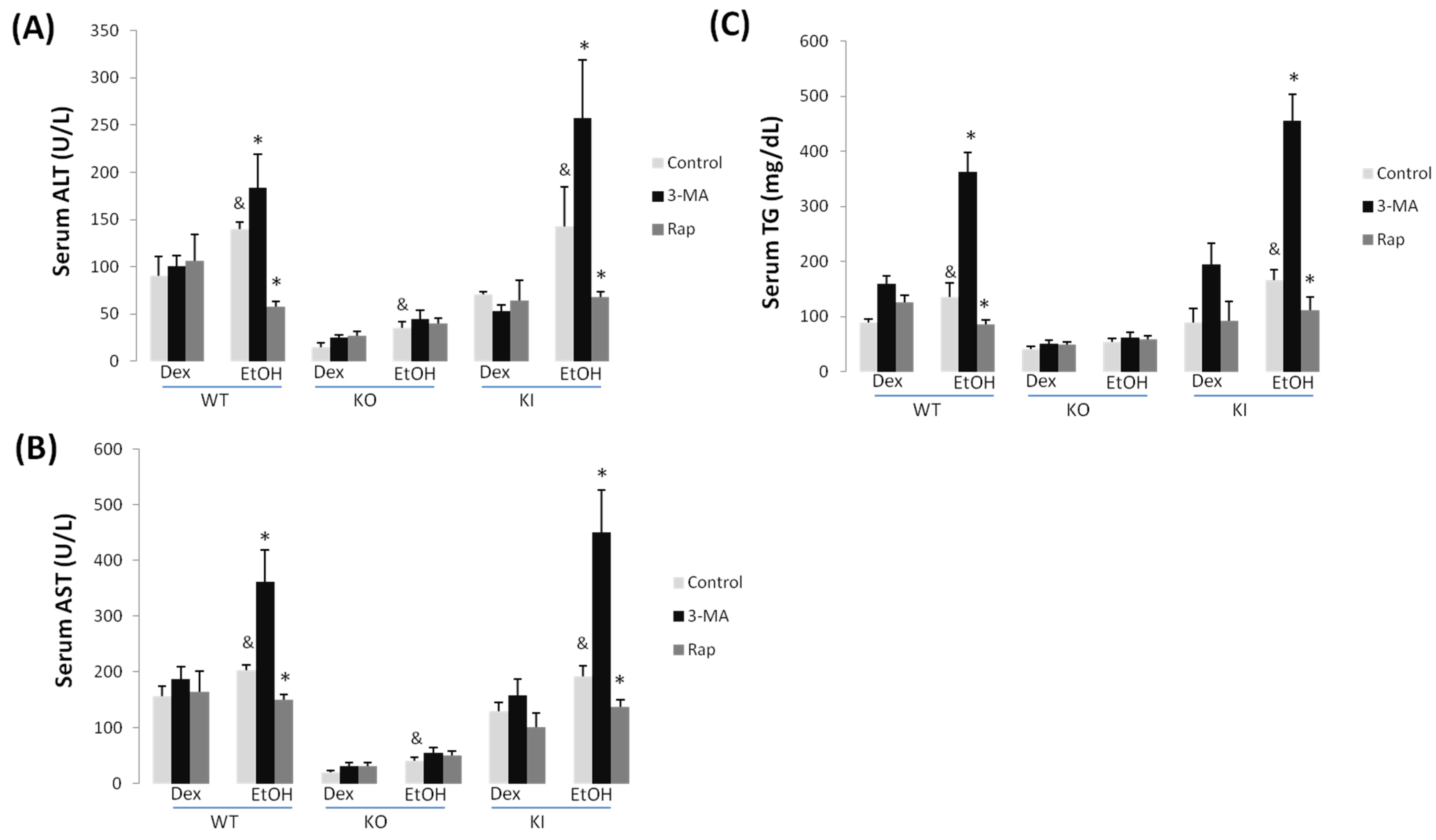
2.1.1. Effect of Chronic Ethanol Treatment on Liver Injury in WT, KO and KI Mice
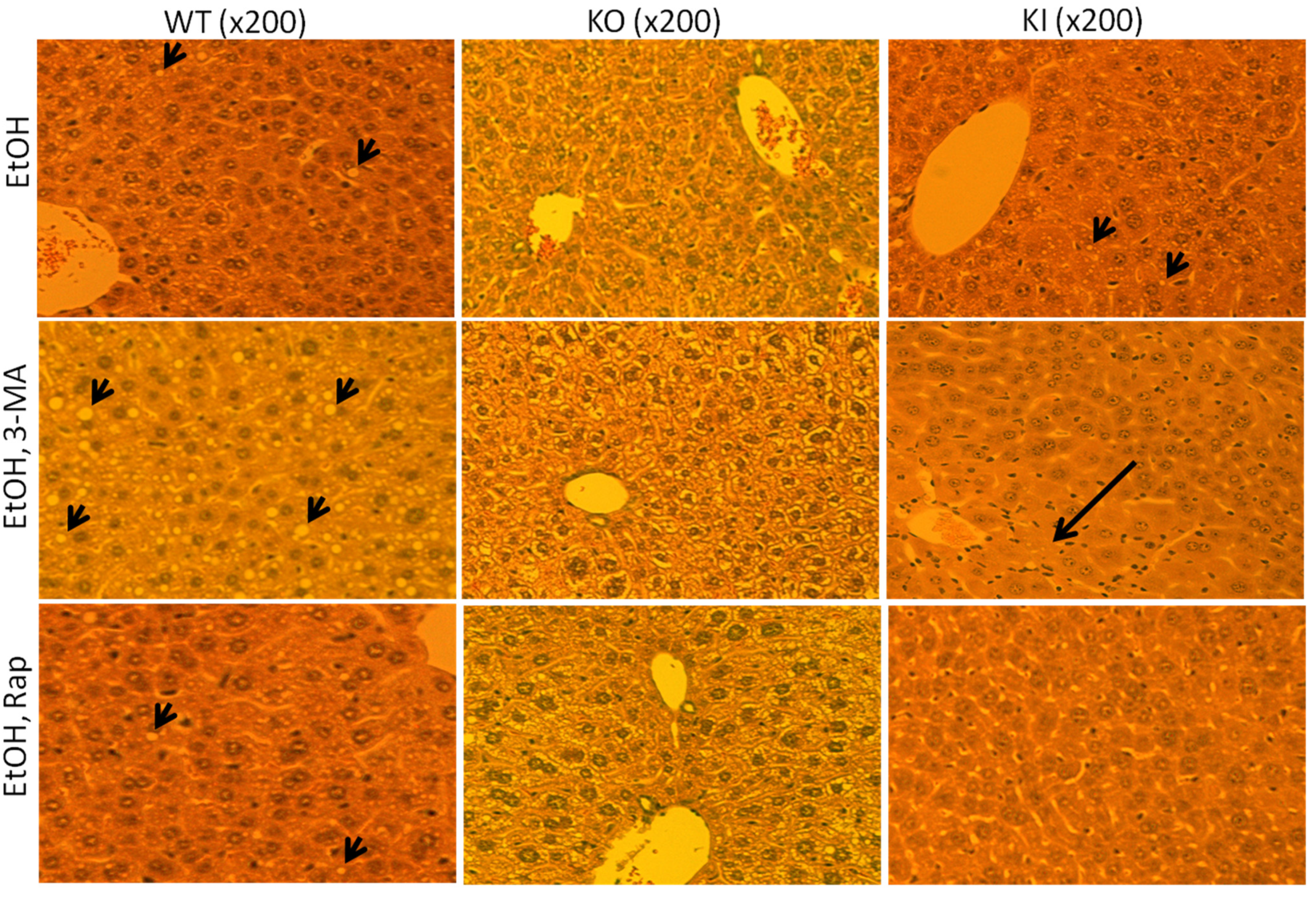
2.1.2. Effect of 3-MA and Rapamycin on Chronic Ethanol-Induced Liver Injury

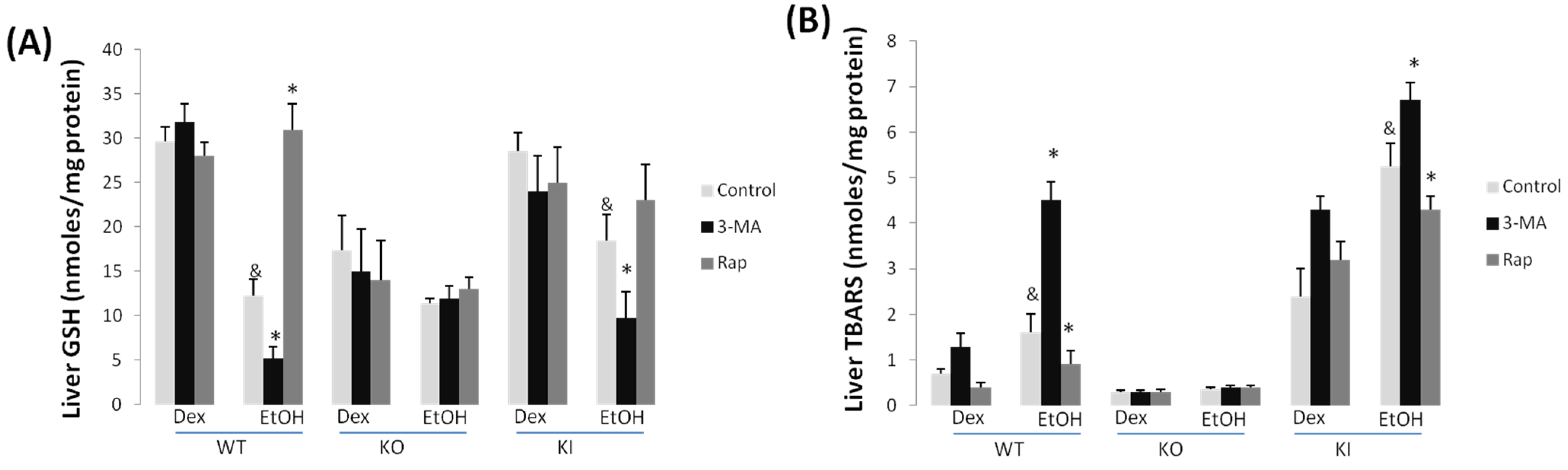
2.1.3. Effect of 3-MA and Rapamycin on Chronic Ethanol-Induced Oxidative Stress and CYP2E1



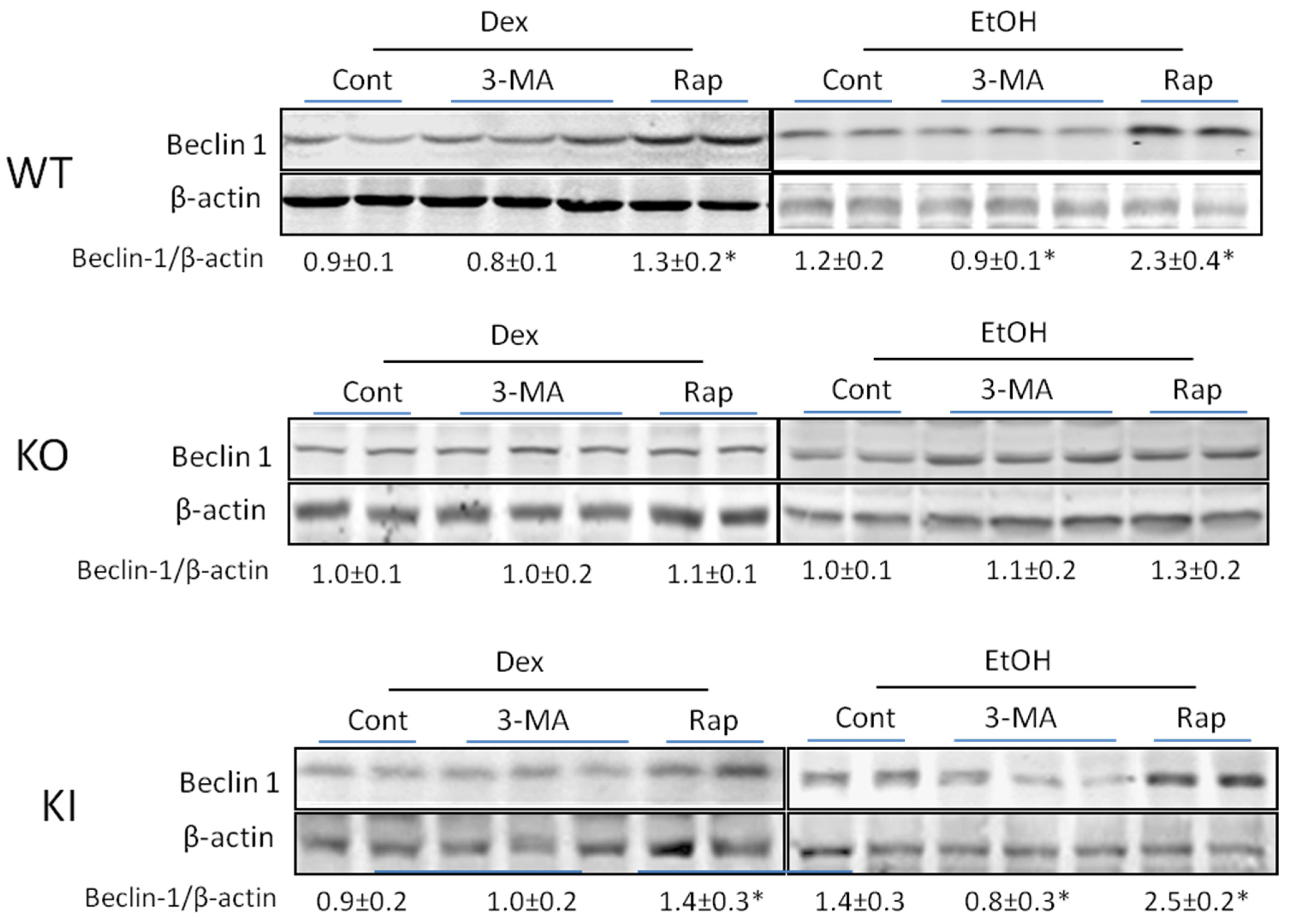
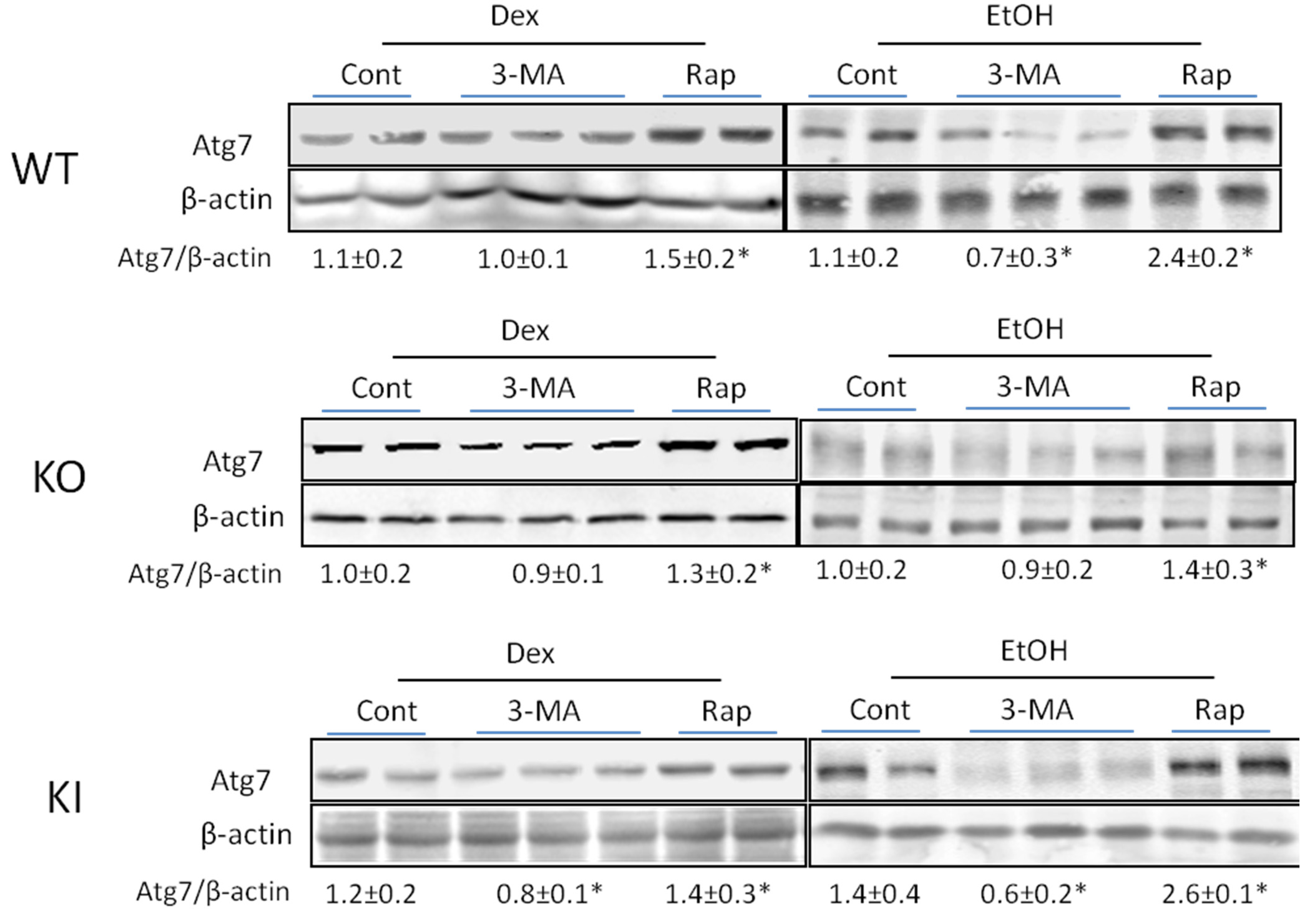
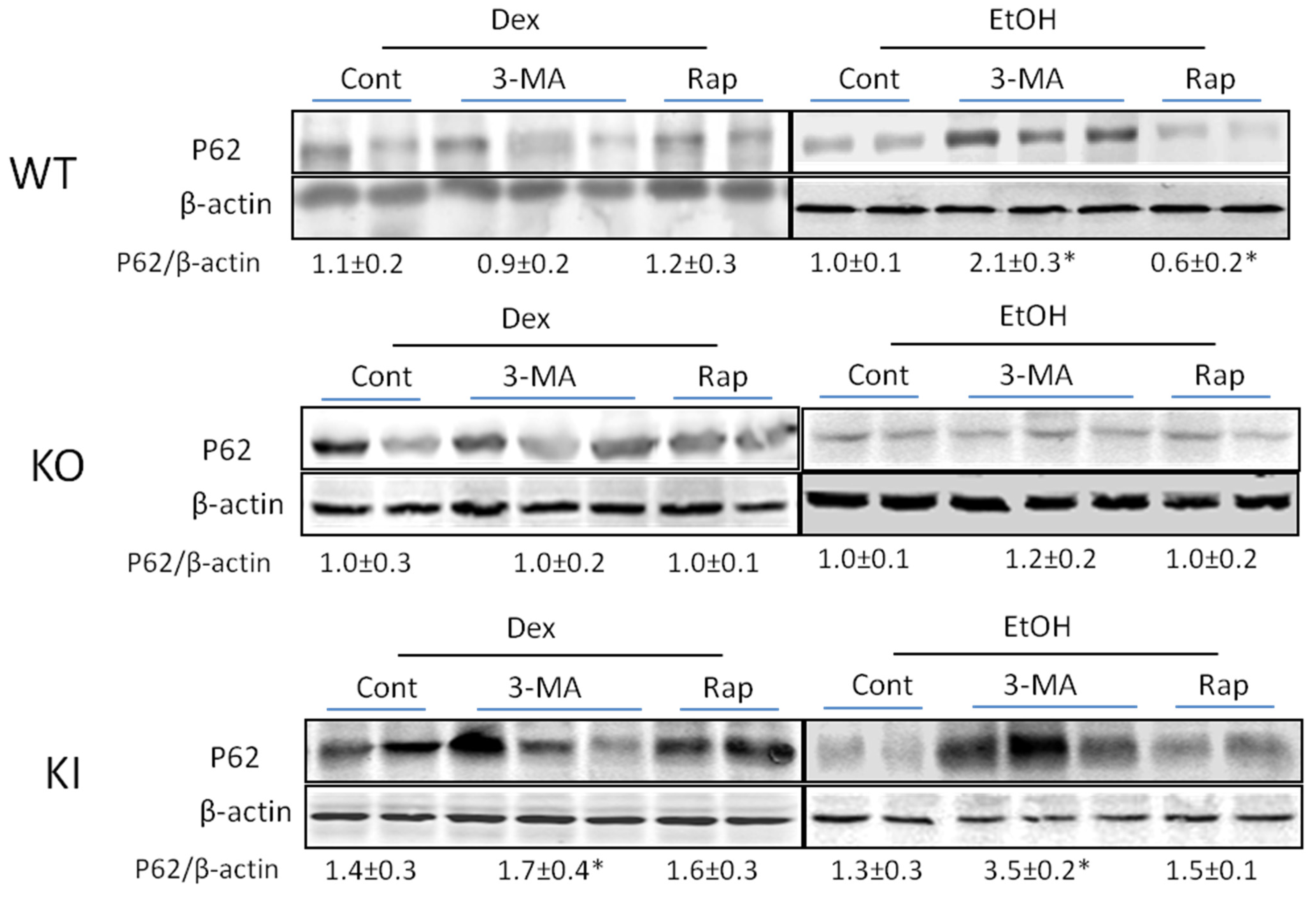
2.1.4. Effects of 3-MA and Rapamycin on Levels of Autophagic Proteins.


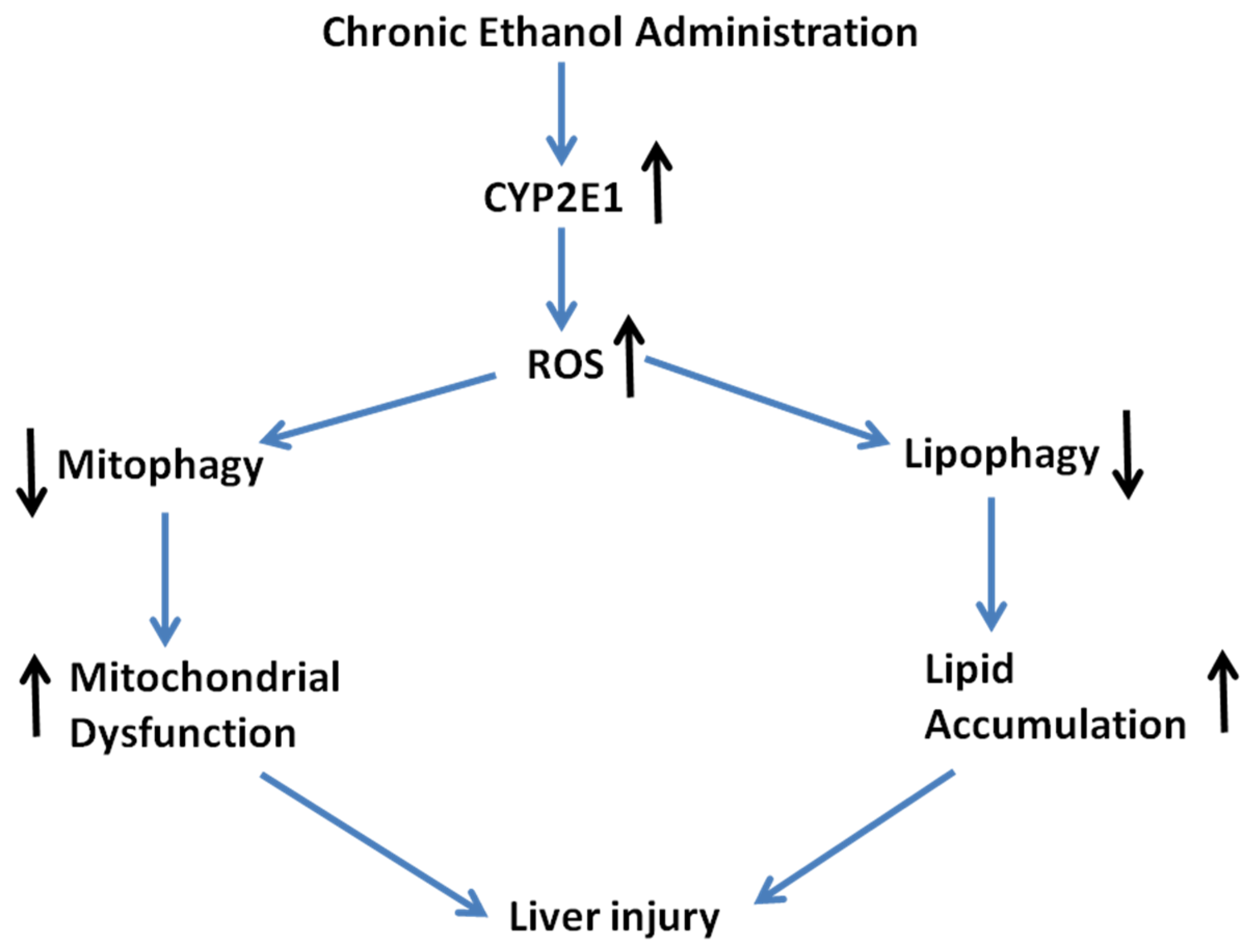
3. Discussion


4. Experimental Section
4.1. Animal Model and Treatment
4.2. Liver Injury Evaluation
4.3. CYP2E1 Activity and Protein Levels
4.4. Levels of Autophagic Proteins
4.5. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Shintanl, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 1, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Manley, S.; Ni, H.N. The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 2011, 236, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Ding, W.X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: Pathophysiological implication. Am. J. Physiol. Cell Physiol. 2010, 298, C973–C978. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Quijano, C.; Wang, J.; Finkel, T. Metabolism meets autophagy. Cell Cycle 2009, 9, 4780–4781. [Google Scholar] [CrossRef]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- French, S.W.; Morimoto, M.; Reitz, R.C.; Koop, D.; Klopfenstein, B.; Estes, K.; Clot, P.; Ingelman-Sundberg, M.; Albano, E. Lipid peroxidation, CYP2E1 and arachidonic acid metabolism in alcoholic liver disease in rats. J. Nutr. 1997, 127, 907S–911S. [Google Scholar] [PubMed]
- Morimoto, M.; Zern, M.A.; Hagbjork, A.L.; Ingelman-Sundberg, M.; French, S.W. Fish oil, alcohol and liver pathology: Role of cytochrome P450 2E1. Proc. Soc. Exp. Biol. Med. 1994, 207, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.A.; Zhao, S.; Sadrzadeh, S.M.H.; Dannenberg, A.J.; Tahan, S.R.; Waxman, D.J. Markedly enhanced cytochrome P4502E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol. Clin. Exp. Res. 1994, 18, 1280–1285. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Hagbjork, A.L.; Wan, Y.J.; Fu, P.C.; Clot, P.; Albano, E.; Ingelman-Sundberg, M.; French, S.W. Modulation of experimental alcohol-induced liver disease by cytochrome P450 2E1 inhibitors. Hepatology 1995, 21, 1610–1617. [Google Scholar] [CrossRef]
- Gouillon, Z.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M., Jr.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Biol. Med. 2000, 224, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Cytochrome p4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol- induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Cederbaum, A.I. Oxidative Stress, Toxicology and Pharmacology of Cyp2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Raucy, J.L.; Kraner, J.C.; Lasker, J.M. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit. Rev. Toxicol. 1993, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.; Koos, P.H.; Their, R. The cytochrome P450 isoenzyme CYP2E1 in the biological processing of industrial chemicals. Int. Arch. Occup. Environ. Health 2003, 76, 174–185. [Google Scholar] [PubMed]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P4502E1. FASEB J. 1992, 6, 724–730. [Google Scholar] [PubMed]
- Song, B.J.; Cederbaum, A.I.; Koop, D.R.; Ingelman-Sundberg, M.; Nanji, A. Ethanol-inducible cytochrome P450 (CYP2E1): Biochemistry, molecular biology and clinical relevance Alcoholism. Clin. Exp. Res. 1996, 20, 138A–146A. [Google Scholar] [CrossRef]
- Tanaka, E.; Terada, M.; Misawa, S. Cytochrome P450 2E1: Its clinical and toxicological role. J. Clin. Pharm. Ther. 2000, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): The first 30 years (1968–1998)—A review. Alcohol. Clin. Exp. Res. 1999, 23, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr. Autophagy and ethanol-induced liver injury. World J. Gastroenterol. 2009, 15, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.N.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Thomes, P.G.; Donohue, T.M., Jr. Involvement of autophagy in alcoholic liver injury and hepatitis c pathogenesis. World J. Gastroenterol. 2011, 17, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Von Haefen, C; Sifringer, M.; Menk, M.; Spies, C.D. Ethanol enhances susceptibility to apoptotic cell death via down regulation of autophagy-related proteins. Alcohol. Clin. Exp. Res. 2011, 35, 1381–1391. [Google Scholar]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and nonalcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Noh, B.; Lee, J.L.; Jun, H.; Lee, H.J.; Jia, Y.; Hoang, M.; Kim, J.; Park, K.; Lee, S. Restoration of autophagy by puerarin in ethanol—Treated hepatocytes via the activation of AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2011, 414, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Thomes, P.G.; Ding, W.X.; Lemasters, J.J.; Donohue, T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcohol. Clin. Exp. Res. 2012, 36, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Thomes, P.C.; Trambly, C.S.; Thiele, G.M.; Duryee, M.J.; Fox, H.S.; Haorah, J.; Donohue, T.M., Jr. Proteosome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem. Biophys. Res. Commun. 2012, 417, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, X.; Zhou, R.; Cederbaum, A.I. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem. Biophys. Res. Commun. 2010, 402, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Inhibition of autophagy promotes CYP2E1-dependent toxicity via elevated oxidative stress, mitochondrial dysfunction and activation of p38 and JNK MAPK. Redox Biol. 2013, 1, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, X.; Zhou, R.; Yang, L.; Cederbaum, A.I. Alcohol steatosis and cytotoxicity: The role of cytochrome P4502E1 and autophagy. Free Radic. Biol. Med. 2012, 53, 1346–1357. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P4502E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Williams, J.A.; Yang, H.; Shi, Y.H.; Fan, J.; Ding, W.X. Targeting autophagy for the treatment of liver diseases. Pharmacol. Res. 2012, 66, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Du, K.D.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, D.; Wang, X.; Cederbaum, A.I. Cytochrome P4502E1, oxidative stress, JNK and autophagy in acute alcohol-induced fatty liver. Free Radic. Biol. Med. 2013, 53, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Veech, R.L.; Park, S.S.; Gelboin, H.V.; Gonzalez, F.J. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J. Biol. Chem. 1989, 264, 3568–3572. [Google Scholar] [PubMed]
- Roberts, B.J.; Shoaf, S.E.; Song, B.J. Rapid changes in cytochrome P4502E1 activity and other P450 enzymes following ethanol withdrawal in rat. Biochem. Pharmacol. 1995, 49, 1665–1673. [Google Scholar] [CrossRef]
- Song, B.J.; Gelboin, H.V.; Park, S.S.; Yang, C.S.; Gonzalez, F.J. Complementary DNA and protein sequences of ethanol-inducible rat and human cytochrome P450s: Transcriptional and posttranscriptional regulation of the rat enzyme. J. Biol. Chem. 1986, 261, 16689–16697. [Google Scholar] [PubMed]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy signaling through reactive oxygen species. Antioxid. Redox Signal. 2011, 14, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Shouvel-Scherz, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Shouvel-Scherz, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Yosef, N.; Ber, Y.; Rubinstein, A.D.; Mor, I.; Sharan, R.; Ruppin, E.; Kimchi, A. A systems level strategy for analyzing the cell death network; implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 2010, 17, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lenardo, M.J. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev. Cell 2007, 12, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic. Biol. Med. 2012, 53, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. 2005, 569, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Nepal, S.; Park, P.H. Activation of autophagy by globular adiponectin attenuates ethanol-inducible apoptosis in HepG2 cells: Involvement of AMPK/FoxO3A axis. Biochim. Biophys. Acta 2013, 1833, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Buter, J.T.; Pineau, T.; Fernandez-Salguero, P.; Gonzalez, F.J. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Yu, A.M.; Ward, J.M.; Krausz, J.W.; Akiyama, T.E.; Feigenbaum, L.; Gonzalez, F.J. The CYP2E1-humanized transgenic mouse: Role of CYP2E1 in acetaminophen hepatoxicity. Drug Metab. Dispos. 2005, 33, 449–457. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Cederbaum, A.I. Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity. Biomolecules 2015, 5, 2659-2674. https://doi.org/10.3390/biom5042659
Lu Y, Cederbaum AI. Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity. Biomolecules. 2015; 5(4):2659-2674. https://doi.org/10.3390/biom5042659
Chicago/Turabian StyleLu, Yongke, and Arthur I. Cederbaum. 2015. "Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity" Biomolecules 5, no. 4: 2659-2674. https://doi.org/10.3390/biom5042659
APA StyleLu, Y., & Cederbaum, A. I. (2015). Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity. Biomolecules, 5(4), 2659-2674. https://doi.org/10.3390/biom5042659