
A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response

 ,
,
Abstract
:1. Introduction
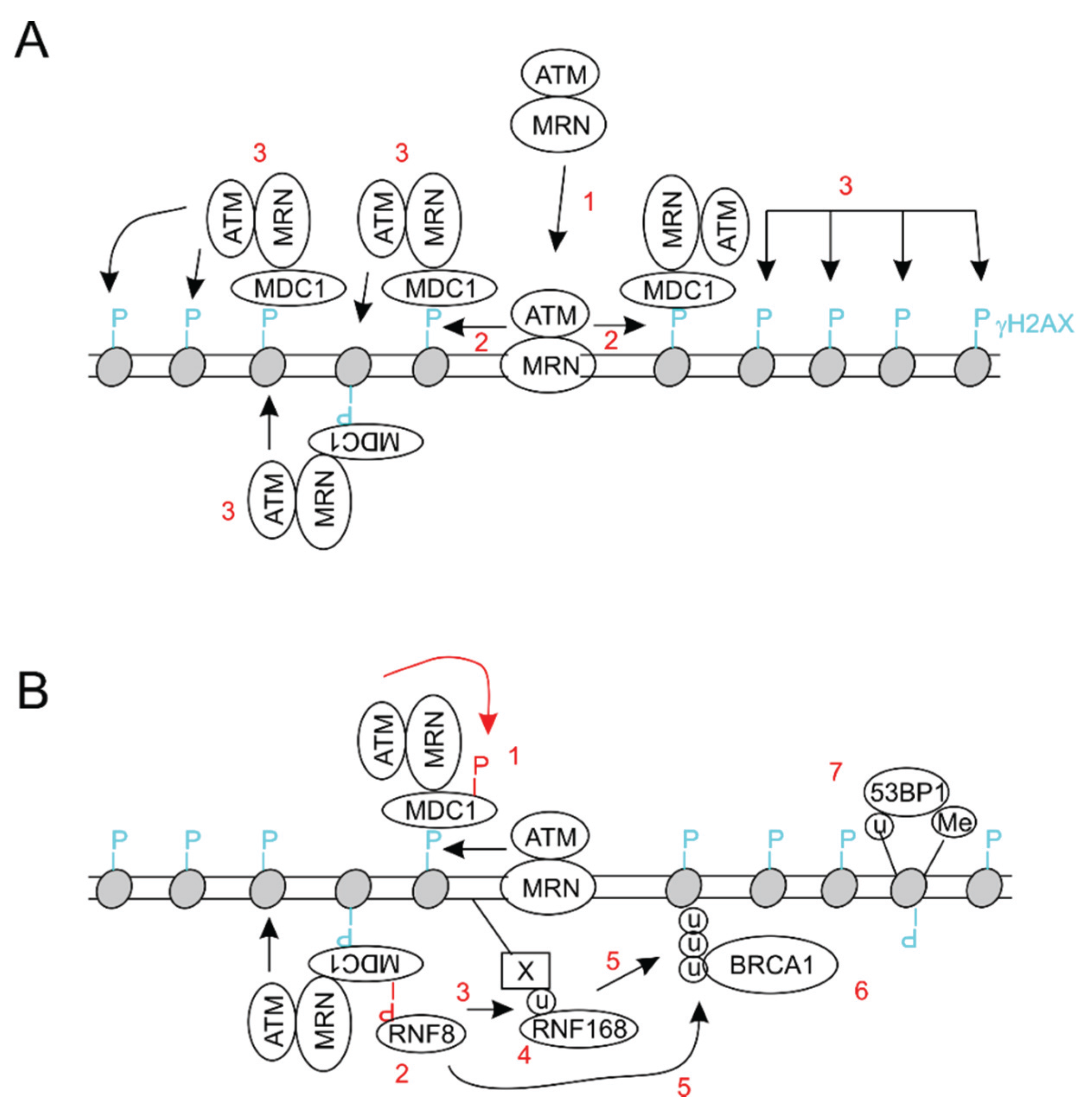
2. General Aspects of DNA Damage Response

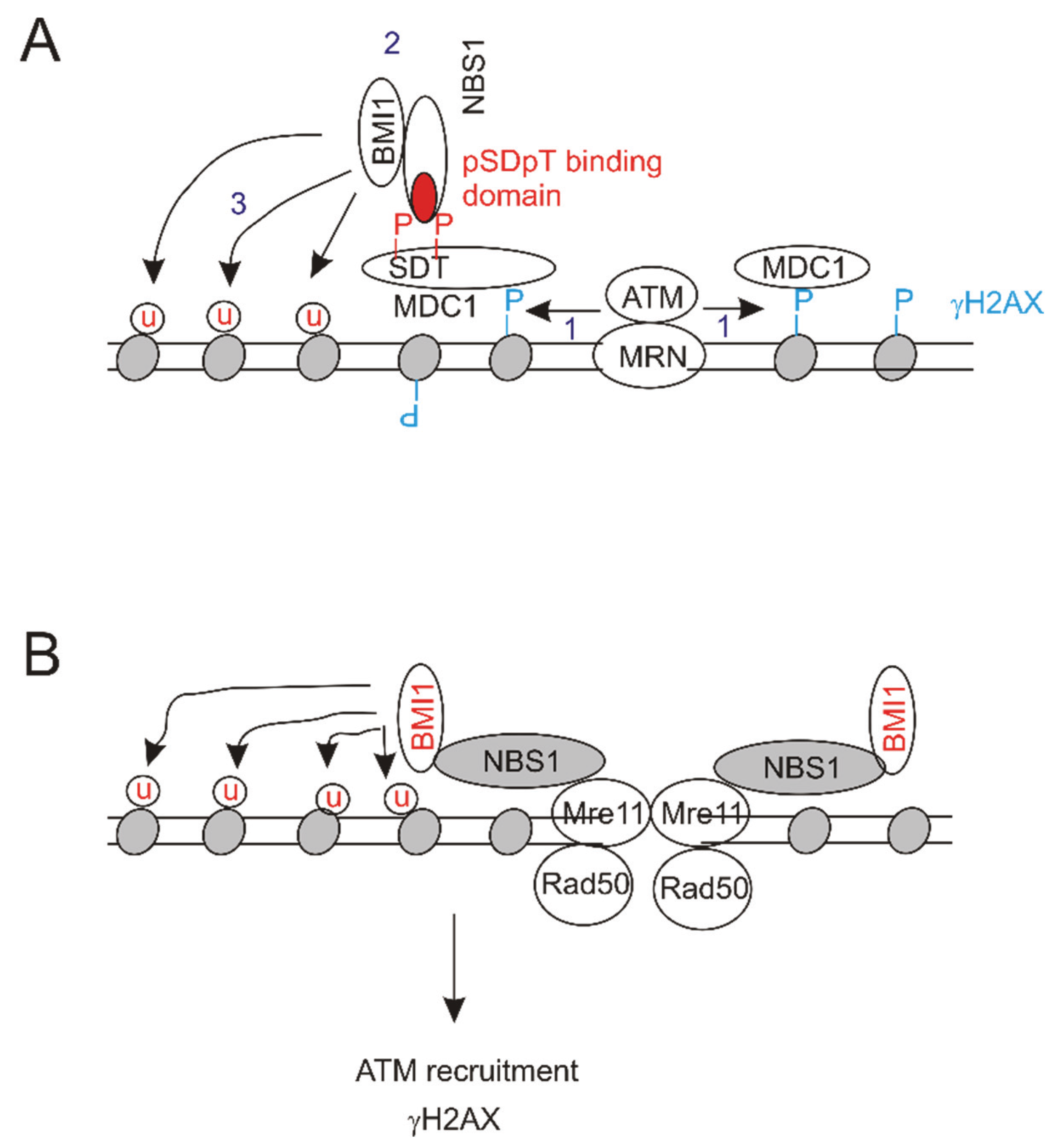
3. BMI1 Enhances DSB Repair by Promoting Histone H2A and γH2AX Ubiquitination
4. The BMI1/RING1b E3 Ubiquitin Ligase Contributes to DDR-Induced Transcription Repression
5. BMI1 Attenuates DSB-Induced Checkpoint Activation by Reducing ATM Activation
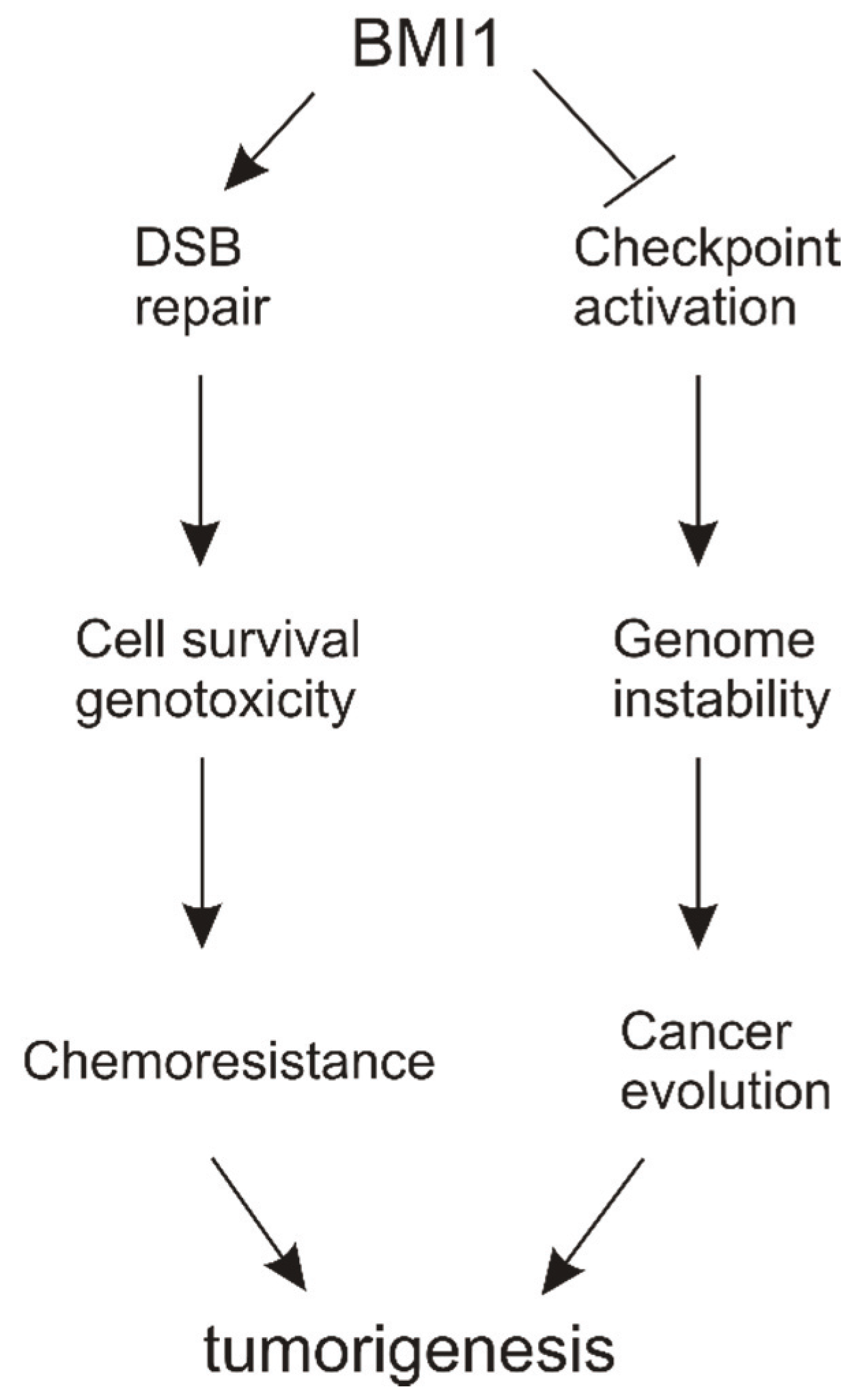
6. Conclusions and Perspectives
6.1. Molecular Mechanisms of BMI1’s Contributions to DSB Repair


{kind=link}
{kind=link}
{kind=link}
| Cell Type | DSB Induction | γH2AX-Mediated BMI1 Recruitment | References |
|---|---|---|---|
| MEFs 1 | UV laser 3 | NO | [74] |
| Hum fib 2 | Ionizing radiation | NO 4 | [75] |
| MEFs | UV laser | YES | [76] |
| HeLa | UV laser | YES 4 | [76] |
| U2OS | Ionizing radiation | BMI1-mediated H2AX Ub 5 enhances γH2AX 6 | [77] |
6.2. Functions of BMI1 in Reducing DDR-Elicited Checkpoint Activation
6.3. DSB Repair vs. Checkpoint Activation

Acknowledgement
Author Contributions
Conflicts of Interest
References
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar] [CrossRef]
- Van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- Cao, R.; Tsukada, Y.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- De Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cao, R.; Wang, M.; Myers, M.P.; Zhang, Y.; Xu, R.M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 20643–20649. [Google Scholar] [CrossRef] [PubMed]
- Leeb, M.; Wutz, A. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J. Cell Biol. 2007, 178, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Yasunaga, S.; Ohno, Y.; Tsumura, M.; Okada, S.; Ishikawa, N.; Shirao, K.; Kikuchi, A.; Nishitani, H.; Kobayashi, M.; et al. Polycomb-group complex 1 acts as an E3 ubiquitin ligase for geminin to sustain hematopoietic stem cell activity. Proc. Natl. Acad. Sci. USA 2008, 105, 10396–10401. [Google Scholar] [CrossRef] [PubMed]
- Bednar, F.; Schofield, H.K.; Collins, M.A.; Yan, W.; Zhang, Y.; Shyam, N.; Eberle, J.A.; Almada, L.L.; Olive, K.P.; Bardeesy, N.; et al. Bmi1 is required for the initiation of pancreatic cancer through an Ink4a-independent mechanism. Carcinogenesis 2015, 36, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Jones, P.A. Tissue-specific alternative splicing in the human INK4a/Arf cell cycle regulatory locus. Oncogene 1999, 18, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Tort, F.; Pinyol, M.; Puig, X.; Hernandez, L.; Hernandez, S.; Fernandez, P.L.; van Lohuizen, M.; Colomer, D.; Campo, E. Bmi-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001, 61, 2409–2412. [Google Scholar] [PubMed]
- Goel, H.L.; Chang, C.; Pursell, B.; Leav, I.; Lyle, S.; Xi, H.S.; Hsieh, C.C.; Adisetiyo, H.; Roy-Burman, P.; Coleman, I.M.; et al. VEGF/neuropilin-2 regulation of Bmi-1 and consequent repression of IGF-IR define a novel mechanism of aggressive prostate cancer. Cancer Dis. 2012, 2, 906–921. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Kuraishy, A.I.; Shalapour, S.; Strasner, A.; Ramirez-Sanchez, C.; Zhang, W.; Shabaik, A.; Karin, M. An IKKalpha-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013, 27, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; He, L.; Kapoor, A.; Gillis, A.; Rybak, A.P.; Cutz, J.C.; Tang, D. Bmi1 promotes prostate tumorigenesis via inhibiting p16(INK4a) and p14(ARF) expression. Biochim. Biophys. Acta 2008, 1782, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Vonlanthen, S.; Heighway, J.; Altermatt, H.J.; Gugger, M.; Kappeler, A.; Borner, M.M.; van Lohuizen, M.; Betticher, D.C. The Bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4a-ARF locus expression. Br. J. Cancer 2001, 84, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yoon, S.Y.; Kim, C.N.; Joo, J.H.; Moon, S.K.; Choe, I.S.; Choe, Y.K.; Kim, J.W. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett. 2004, 203, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yoon, S.Y.; Jeong, S.H.; Kim, S.Y.; Moon, S.K.; Joo, J.H.; Lee, Y.; Choe, I.S.; Kim, J.W. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast 2004, 13, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Song, L.B.; Zeng, M.S.; Liao, W.T.; Zhang, L.; Mo, H.Y.; Liu, W.L.; Shao, J.Y.; Wu, Q.L.; Li, M.Z.; Xia, Y.F.; et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006, 66, 6225–6232. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Bath, M.L.; Harris, A.W.; Adams, J.M. Bmi-1 transgene induces lymphomas and collaborates with Myc in tumorigenesis. Oncogene 1993, 8, 3161–3164. [Google Scholar] [PubMed]
- Alkema, M.J.; Jacobs, H.; van Lohuizen, M.; Berns, A. Pertubation of B and T cell development and predisposition to lymphomagenesis in Emu Bmi1 transgenic mice require the Bmi1 ring finger. Oncogene 1997, 15, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hu, B.; Li, T.; Ma, J.; Alam, G.; Gunning, W.T.; Ding, H.F. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am. J. Pathol. 2007, 170, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Ferretti, R.; Hilgendorf, K.I.; Perret, C.; Whyte, P.; Lees, J.A. Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene 2014, 33, 3742–3747. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16INK4a and p19ARF senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.; Valk-Lingbeek, M.E.; van der Stoop, P.P.; Jacobs, J.J.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. INK4a and ARF differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Iwama, A.; Morita, Y.; Kamijo, T.; van Lohuizen, M.; Nakauchi, H. Differential impact of INK4a and ARF on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J. Exp. Med. 2006, 203, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers Bmi1 and LGR5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Siddique, H.R.; Saleem, M. Role of Bmi1, a stem cell factor, in cancer recurrence and chemoresistance: Preclinical and clinical evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Zacharek, S.J.; Fillmore, C.M.; Lau, A.N.; Gludish, D.W.; Chou, A.; Ho, J.W.; Zamponi, R.; Gazit, R.; Bock, C.; Jager, N.; et al. Lung stem cell self-renewal relies on Bmi1-dependent control of expression at imprinted loci. Cell Stem Cell 2011, 9, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Dimos, J.T.; Ivanova, N.B.; Lowry, N.; Lemischka, I.R.; Temple, S. shRNA knockdown of Bmi-1 reveals a critical role for p21-RB pathway in NSC self-renewal during development. Cell Stem Cell 2007, 1, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Arribillaga, E.; Rodilla, V.; Pellegrinet, L.; Guiu, J.; Iglesias, M.; Roman, A.C.; Gutarra, S.; Gonzalez, S.; Munoz-Canoves, P.; Fernandez-Salguero, P.; et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of notch. Development 2015, 142, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.; Hulsman, D.; Tanger, E.; Buckle, T.; Blom, M.; Zevenhoven, J.; van Tellingen, O.; van Lohuizen, M. Bmi1 controls tumor development in an INK4a/ARF-independent manner in a mouse model for glioma. Cancer Cell 2007, 12, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.R.; Lee, S.; Ho, C.; Bommi, P.; Huang, S.A.; Cheung, S.T.; Dimri, G.P.; Chen, X. Bmi1 functions as an oncogene independent of INK4a/ARF repression in hepatic carcinogenesis. Mol. Cancer Res. 2009, 7, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Ding, J.; Perdue, A.; Yang, L.; Zha, Y.; Ren, M.; Huang, S.; Cui, H.; Ding, H.F. Cyclin E1 is a common target of BMI1 and MYCN and a prognostic marker for neuroblastoma progression. Oncogene 2012, 31, 3785–3795. [Google Scholar] [CrossRef] [PubMed]
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein BMI-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. BMI1 is essential in twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Martinez, J.L.; Jacobs, J.J.; Keblusek, P.; Itahana, K.; van Lohuizen, M.; Campisi, J.; Wazer, D.E.; Band, V. The BMI-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002, 62, 4736–4745. [Google Scholar] [PubMed]
- Fan, C.; He, L.; Kapoor, A.; Rybak, A.P.; de Melo, J.; Cutz, J.C.; Tang, D. PTEN inhibits BMI1 function independently of its phosphatase activity. Mol. Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Polycomb group proteins and myc: The cancer connection. Cell. Mol. Life Sci. 2014, 71, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-CHK2 and ATR-CHK1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 2010, 9, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Jackson, S.P. MDC1/NFBD1: A key regulator of the DNA damage response in higher eukaryotes. DNA Repair 2004, 3, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Spycher, C.; Miller, E.S.; Townsend, K.; Pavic, L.; Morrice, N.A.; Janscak, P.; Stewart, G.S.; Stucki, M. Constitutive phosphorylation of MDC1 physically links the Mre11-Rad50-Nbs1 complex to damaged chromatin. J. Cell Biol. 2008, 181, 227–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the Mrn complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. Mdc1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Pinato, S.; Scandiuzzi, C.; Arnaudo, N.; Citterio, E.; Gaudino, G.; Penengo, L. RNF168, a new ring finger, miu-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The riddle syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sakai, W.; Kawamoto, T.; Bree, R.T.; Lowndes, N.F.; Takeda, S.; Taniguchi, Y. Genetic dissection of vertebrate 53bp1: A major role in non-homologous end joining of DNA double strand breaks. DNA Repair 2006, 5, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53bp1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53bp1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53bp1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. Bmi1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. Bmi1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. Bmi1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Peng, G.; Hung, W.C.; Lin, S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 2011, 286, 28599–28607. [Google Scholar] [CrossRef] [PubMed]
- Chagraoui, J.; Hebert, J.; Girard, S.; Sauvageau, G. An anticlastogenic function for the polycomb group gene BMI1. Proc. Natl. Acad. Sci. USA 2011, 108, 5284–5289. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yan, J.; Tang, D. ERK kinases modulate the activation of PI3 kinase related kinases (PIKKs) in DNA damage response. Histol. Histopathol. 2013, 28, 1547–1554. [Google Scholar] [PubMed]
- Bartocci, C.; Diedrich, J.K.; Ouzounov, I.; Li, J.; Piunti, A.; Pasini, D.; Yates, J.R., 3rd; Lazzerini Denchi, E. Isolation of chromatin from dysfunctional telomeres reveals an important role for Ring1B in NHEJ-mediated chromosome fusions. Cell Rep. 2014, 7, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Gieni, R.S.; Ismail, I.H.; Campbell, S.; Hendzel, M.J. Polycomb group proteins in the DNA damage response: A link between radiation resistance and “stemness”. Cell Cycle 2011, 10, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, C.S.; Halligan, B.W.; Levy, L.S. The Bmi-1 oncoprotein interacts with ding and MPH2: The role of ring finger domains. Oncogene 1998, 16, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Alpert, A.; Leiter, C.; Gong, F.; Jackson, S.P.; Miller, K.M. Systematic identification of functional residues in mammalian histone H2AX. Mol. Cell. Biol. 2013, 33, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.; Agarwal, P.; Canny, M.D.; Gong, F.; Robison, A.D.; Finkelstein, I.J.; Durocher, D.; Miller, K.M. Nucleosome acidic patch promotes RNF168- and Ring1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Henrici, R.C.; Tan, S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 2014, 514, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for pbaf in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Nacerddine, K.; Beaudry, J.B.; Ginjala, V.; Westerman, B.; Mattiroli, F.; Song, J.Y.; van der Poel, H.; Ponz, O.B.; Pritchard, C.; Cornelissen-Steijger, P.; et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J. Clin. Investig. 2012, 122, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. Brca1 recruitment to transcriptional pause sites is required for R-Loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Nagaura, Y.; Yasui, A. Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 2015, 58, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of CDC25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the CHK1 checkpoint pathway in mammals: Linkage of DNA damage to CDK regulation through CDC25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Hosing, A.S.; Kundu, S.T.; Dalal, S.N. 14-3-3 gamma is required to enforce both the incomplete S phase and G2 DNA damage checkpoints. Cell Cycle 2008, 7, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Oh, J.E.; Chen, W.; Kim, R.; Kim, R.H.; Shin, K.H.; McBride, W.H.; Park, N.H.; Kang, M.K. Radioprotective effects of BMI-1 involve epigenetic silencing of oxidase genes and enhanced DNA repair in normal human keratinocytes. J. Investig. Dermatol. 2011, 131, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Bhattacharyya, S.; Szabolcs, A.; Rodriguez-Aguayo, C.; Jennings, N.B.; Lopez-Berestein, G.; Mukherjee, P.; Sood, A.K.; Bhattacharya, R. Enhancing chemotherapy response with BMI-1 silencing in ovarian cancer. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ojo, D.; Lin, X.; Wong, N.; He, L.; Yan, J.; Xu, S.; Major, P.; Tang, D. Bmi1 attenuates etoposide-induced G2/M checkpoints via reducing ATM activation. Oncogene 2015, 34, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.T.; Schwartz, R.A.; Stracker, T.H.; Lilley, C.E.; Lee, D.V.; Weitzman, M.D. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22, 6610–6620. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Atm activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Goldknopf, I.L.; Busch, H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. USA 1977, 74, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Hunt, L.T.; Dayhoff, M.O. Amino-terminal sequence identity of ubiquitin and the nonhistone component of nuclear protein A24. Biochem. Biophys. Res. Commun. 1977, 74, 650–655. [Google Scholar] [CrossRef]
- Buchwald, G.; van der Stoop, P.; Weichenrieder, O.; Perrakis, A.; van Lohuizen, M.; Sixma, T.K. Structure and E3-ligase activity of the ring-ring complex of polycomb proteins Bmi1 and Ring1B. EMBO J. 2006, 25, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.; Chapman, J.R.; Clapperton, J.A.; Haire, L.F.; Hartsuiker, E.; Li, J.; Carr, A.M.; Jackson, S.P.; Smerdon, S.J. A supramodular FHA/BRCT-repeat architecture mediates NBS1 adaptor function in response to DNA damage. Cell 2009, 139, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hari, F.J.; Spycher, C.; Jungmichel, S.; Pavic, L.; Stucki, M. A divalent FHA/BRCT-binding mechanism couples the Mre11-Rad50-Nbs1 complex to damaged chromatin. EMBO Rep. 2010, 11, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Kang, H.Y.; Yang, W.L.; Wu, J.; Jeong, Y.S.; Wang, J.; Chan, C.H.; Lee, S.W.; Zhang, X.; Lamothe, B.; et al. Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J. Biol. Chem. 2011, 286, 30806–30815. [Google Scholar] [CrossRef] [PubMed]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Becker, E.; Meyer, V.; Madaoui, H.; Guerois, R. Detection of a tandem BRCT in NBS1 and XRS2 with functional implications in the DNA damage response. Bioinformatics 2006, 22, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers CTP1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Desai-Mehta, A.; Cerosaletti, K.M.; Concannon, P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol. Cell. Biol. 2001, 21, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Cheung, N.K.; Vider, J.; Cheung, I.Y.; Gerald, W.L.; Tickoo, S.K.; Holland, E.C.; Blasberg, R.G. Mycn and myc regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. FASEB J. 2011, 25, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
- Waldron, T.; de Dominici, M.; Soliera, A.R.; Audia, A.; Iacobucci, I.; Lonetti, A.; Martinelli, G.; Zhang, Y.; Martinez, R.; Hyslop, T.; et al. c-Myb and its target Bmi1 are required for p190BCR/ABl leukemogenesis in mouse and human cells. Leukemia 2012, 26, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Liu, G.H.; Zhang, H.; Xing, S.; Hu, L.J.; Zhao, W.F.; Xie, B.; Li, M.Z.; Zeng, B.H.; Li, Y.; et al. SP1 and c-Myc regulate transcription of Bmi1 in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2929–2944. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hoenerhoff, M.J.; Bommi, P.; Sainger, R.; Guo, W.J.; Dimri, M.; Band, H.; Band, V.; Green, J.E.; Dimri, G.P. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007, 67, 10286–10295. [Google Scholar] [CrossRef] [PubMed]
- Hoenerhoff, M.J.; Chu, I.; Barkan, D.; Liu, Z.Y.; Datta, S.; Dimri, G.P.; Green, J.E. Bmi1 cooperates with H-Ras to induce an aggressive breast cancer phenotype with brain metastases. Oncogene 2009, 28, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Tatrai, P.; Szepesi, A.; Matula, Z.; Szigeti, A.; Buchan, G.; Madi, A.; Uher, F.; Nemet, K. Combined introduction of Bmi-1 and hTERT immortalizes human adipose tissue-derived stromal cells with low risk of transformation. Biochem. Biophys. Res. Commun. 2012, 422, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Dakic, A.; Chen, R.; Palechor-Ceron, N.; Dai, Y.; Kallakury, B.; Schlegel, R.; Liu, X. HPV16 E7 protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Chen, Z.; Hu, F.; Tao, Q.; Lam, A.K. Bmi-1 activation is crucial in hTERT-induced epithelial-mesenchymal transition of oral epithelial cells. Experimental Mol. Pathol. 2013, 95, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.P.; Peddibhotla, S.; Bu, W.; Zhao, J.; Haricharan, S.; Du, Y.C.; Podsypanina, K.; Rosen, J.M.; Donehower, L.A.; Li, Y. Defining the ATM-mediated barrier to tumorigenesis in somatic mammary cells following ErbB2 activation. Proc. Natl. Acad. Sci. USA 2010, 107, 3728–3733. [Google Scholar] [CrossRef] [PubMed]
- Takacova, S.; Slany, R.; Bartkova, J.; Stranecky, V.; Dolezel, P.; Luzna, P.; Bartek, J.; Divoky, V. DNA damage response and inflammatory signaling limit the MLL-ENL-induced leukemogenesis in vivo. Cancer Cell 2012, 21, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Amati, B. Two sides of the myc-induced DNA damage response: From tumor suppression to tumor maintenance. Cell Div. 2012. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D. A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response. Biomolecules 2015, 5, 3396-3415. https://doi.org/10.3390/biom5043396
Lin X, Ojo D, Wei F, Wong N, Gu Y, Tang D. A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response. Biomolecules. 2015; 5(4):3396-3415. https://doi.org/10.3390/biom5043396
Chicago/Turabian StyleLin, Xiaozeng, Diane Ojo, Fengxiang Wei, Nicholas Wong, Yan Gu, and Damu Tang. 2015. "A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response" Biomolecules 5, no. 4: 3396-3415. https://doi.org/10.3390/biom5043396
APA StyleLin, X., Ojo, D., Wei, F., Wong, N., Gu, Y., & Tang, D. (2015). A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response. Biomolecules, 5(4), 3396-3415. https://doi.org/10.3390/biom5043396