
Activation of the DNA Damage Response by RNA Viruses

Abstract
:1. RNA Viruses
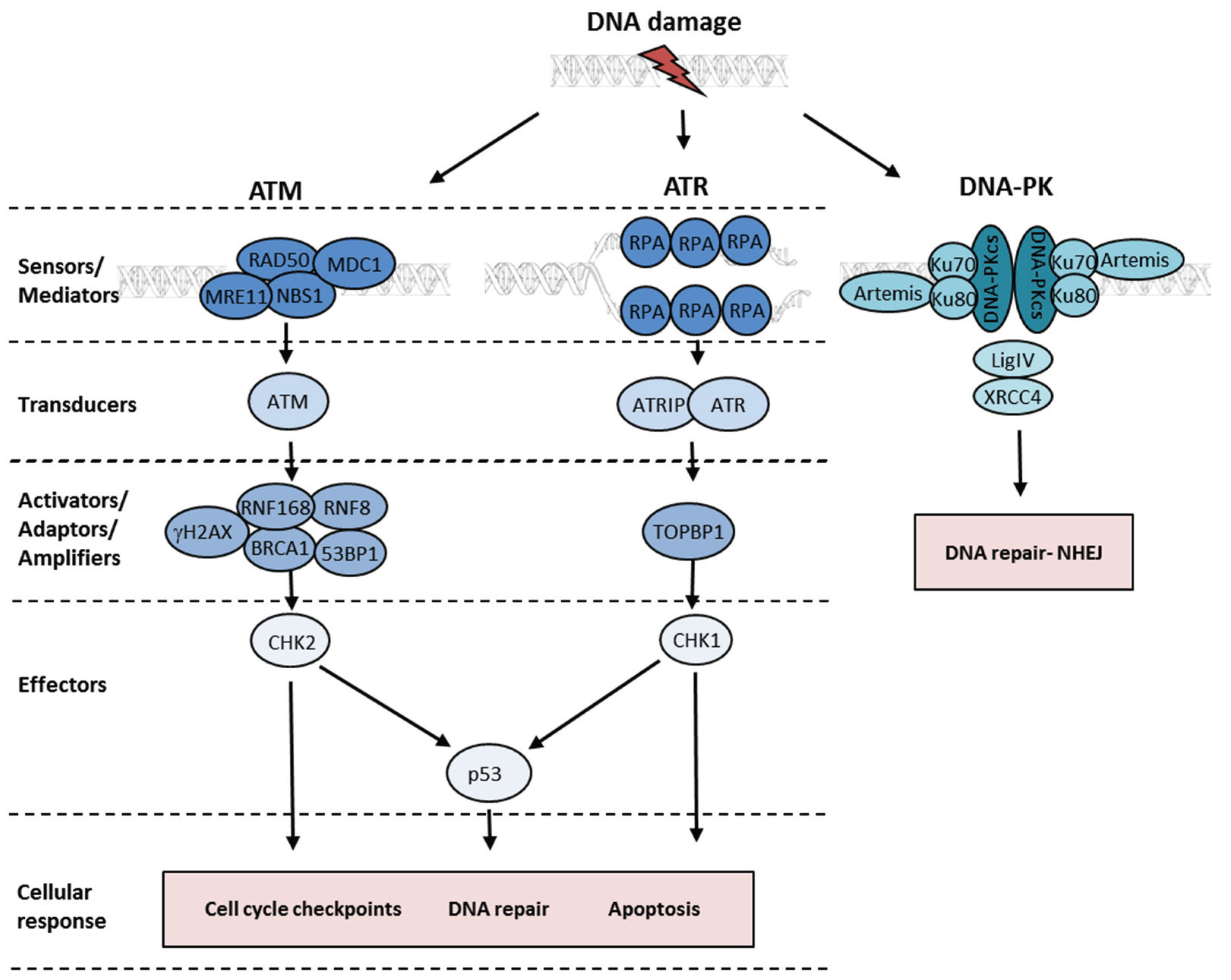
2. The Cellular DNA Damage Response and DNA Repair Pathways

3. The ATM Kinase and the Detection of Double-Strand Breaks
4. ATR Kinase Activation at Single-Stranded DNA
5. The DDR and Cell Cycle Checkpoints
6. Double-Strand Break Repair
7. Repair of Single-Strand DNA Damage
8. DNA Damage and Apoptosis
9. Non-Coding RNAs and the DDR
10. Viral Interaction with the DDR
11. RNA Viruses and DNA Damage

{kind=link}
{kind=link}
| Virus | Family | RNA Genome Conformation | DDR Consequences | Representative References |
|---|---|---|---|---|
| Human immunodeficiency Virus 1 (HIV-1) | Retroviridae | + single strand (2 copies) | Activation of ATR, replication stress, activation of nucleases and formation of DDR foci by Vpr | [92,93,94,95] |
| Human T-cell lymphotropic virus 1 (HTLV-1) | Retroviridae | + single strand (2 copies) | Genome instability and DNA damage; attenuation of BER, NER, MMR, HR, NHEJ pathways by Tax. Generation of ROS | [96,97,98] |
| Hepatitis C virus (HCV) | Flaviviridae | + single strand | Generation of ROS and NO, reduced expression of MMR, BER and NER components, interaction of viral proteins with ATM and modulation of ATM pathway activity | [99,100,101,102,103] |
| Infectious bronchitis virus (IBV) | Coronaviridae | + single strand | Activation of ATR pathway and DNA replication stress | [104] |
| Influenza A virus | Orthomyxoviridae | − single strand | Direct DNA damage (comet assay) Induction of γH2AX foci | [105,106] |
| Chikungunya virus | Togaviridae | + single strand | Induction of GADD34 expression | [107] |
| Sindbis virus | Togaviridae | + single strand | Activation of PARP-1 | [108] |
| La Crosse virus | Bunyaviridae | − single strand | Increased phosphorylation of H2AX | [109] |
| Rift valley fever virus (RVFV) | Bunyaviridae | − single strand | Activation of ATM signalling; inhibition of ATR | [110] |
| Avian Reovirus (ARV) | Reoviridae | double strand | Genome instability and activation of ATR signalling | [111] |
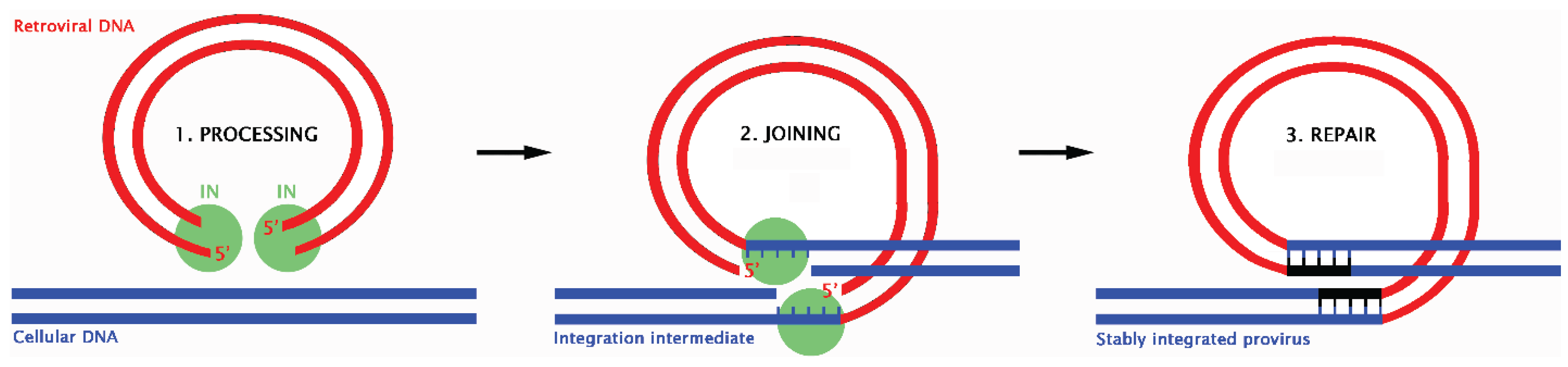
12. Retroviral Integration and the DDR

13. Human Immunodeficiency Virus 1 (HIV-1) Vpr Protein
14. Human T-Cell Lymphotropic Virus Type 1 (HTLV-1) Tax Protein
15. Hepatitis C Virus (HCV)
16. Infectious Bronchitis Virus (IBV)
17. Influenza A Virus
18. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tao, Y.J.; Ye, Q. RNA virus replication complexes. PLoS Pathog. 2010, 6, e1000943. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Belshaw, R.; Pybus, O.G.; Rambaut, A. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res. 2007, 17, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.R. Seeking membranes: Positive-strand RNA virus replication complexes. PLoS Biol. 2008, 6, e270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, Y.; Guo, Y.; Lou, Z. Structural perspective on the formation of ribonucleoprotein complex in negative-sense single-stranded RNA viruses. Trends Microbiol. 2013, 21, 475–484. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.; Summers, M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005, 3, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.P.; Grandgenett, D.P. Genetic evidence that the avian retrovirus DNA endonuclease domain of pol is necessary for viral integration. J. Virol. 1988, 62, 2307–2312. [Google Scholar] [PubMed]
- Caly, L.; Wagstaff, K.M.; Jans, D.A. Nuclear trafficking of proteins from RNA viruses: Potential target for antivirals? Antiviral.Res. 2012, 95, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [PubMed]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell Biol. 2001, 13, 738–747. [Google Scholar] [CrossRef]
- Costanzo, V.; Robertson, K.; Ying, C.Y.; Kim, E.; Avvedimento, E.; Gottesman, M.; Grieco, D.; Gautier, J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell 2000, 6, 649–659. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Petrini, J.H.; Williams, B.R.; Lukas, J.; Bartek, J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002, 30, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.M.; Ferris, D.K.; Hoffmann, I.; Jackman, J.; Draetta, G.; Kohn, K.W. Role of the cdc25C phosphatase in G2 arrest induced by nitrogen mustard. Proc. Natl. Acad. Sci. USA 1994, 91, 9480–9484. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007. [Google Scholar] [CrossRef] [PubMed]
- Suwa, A.; Hirakata, M.; Takeda, Y.; Jesch, S.A.; Mimori, T.; Hardin, J.A. DNA-dependent protein-kinase (Ku Protein-P350 Complex) assembles on double-stranded DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 6904–6908. [Google Scholar] [CrossRef] [PubMed]
- Leber, R.; Wise, T.W.; Mizuta, R.; Meek, K. The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J. Biol. Chem. 1998, 273, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Suzuki, N.; Namba, N.; Umeda, N.; Ma, X.J.; Morita, A.; Tomita, M.; Enomoto, A.; Serizawa, S.; Hirano, K.; et al. Cleavage and phosphorylation of XRCC4 protein induced by X-irradiation. FEBS Lett. 2000, 478, 67–71. [Google Scholar] [CrossRef]
- Calsou, P.; Delteil, C.; Frit, P.; Droulet, J.; Salles, B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol. 2003, 326, 93–103. [Google Scholar] [CrossRef]
- Lieber, M.R.; Gu, J.; Lu, H.; Shimazaki, N.; Tsai, A.G. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell. Biochem. 2010, 50, 279–296. [Google Scholar] [PubMed]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Shinagawa, H.; Iwasaki, H. Processing the holliday junction in homologous recombination. Trends Biochem. Sci. 1996, 21, 107–111. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1262. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Sung, J.S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, A.E.; Chen, L.; Dong, Z.; Leppard, J.B.; Levin, D.S.; Mackey, Z.B.; Motycka, T.A. Completion of base excision repair by mammalian DNA ligases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 151–164. [Google Scholar] [PubMed]
- Wood, R.D. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 1999, 81, 39–44. [Google Scholar] [CrossRef]
- Graf, N.; Ang, W.H.; Zhu, G.; Myint, M.; Lippard, S.J. Role of endonucleases XPF and XPG in nucleotide excision repair of platinated DNA and cisplatin/oxaliplatin cytotoxicity. Chembiochem 2011, 12, 1115–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.F.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.K.; Kenny, M.K.; Wood, R.D. Proliferating cell nuclear antigen is required for DNA excision repair. Cell 1992, 69, 367–374. [Google Scholar] [CrossRef]
- Drummond, J.T.; Li, G.M.; Longley, M.J.; Modrich, P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor-cells. Science 1995, 268, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Palombo, F.; Iaccarino, I.; Nakajima, E.; Ikejima, M.; Shimada, T.; Jiricny, J. hMutSβ, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion leaps in DNA. Curr. Biol. 1996, 6, 1181–1184. [Google Scholar] [CrossRef]
- McCulloch, S.D.; Gu, L.Y.; Li, G.M. Bi-directional processing of DNA loops by mismatch repair-dependent and -independent pathways in human cells. J. Biol. Chem. 2003, 278, 3891–3896. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Y.; Hong, Y.; McCulloch, S.; Watanabe, H.; Li, G.M. ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res. 1998, 26, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Pierce, A.J.; Modrich, P. DNA polymerase delta is required for human mismatch repair in vitro. J. Biol. Chem. 1997, 272, 10917–10921. [Google Scholar] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Suzuki, S.; Okamoto, H.; Yoshida, H.; Ohba, Y.; Takaoka, A.; Taniguchi, T. Differential contribution of Puma and Noxa in dual regulation of p53-mediated apoptotic pathways. EMBO J. 2006, 25, 4952–4962. [Google Scholar] [CrossRef] [PubMed]
- Jacotot, E.; Ravagnan, L.; Loeffler, M.; Ferri, K.F.; Vieira, H.L.; Zamzami, N.; Costantini, P.; Druillennec, S.; Hoebeke, J.; Briand, J.P.; et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000, 191, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Cherry, S.; Dybdahl-Sissoko, N.; Neumann, G.; Kawaoka, Y.; Hinshaw, V.S. Influenza virus NS1 protein induces apoptosis in cultured cells. J. Virol. 2001, 75, 7875–7881. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- You, L.R.; Chen, C.M.; Lee, Y.H. Hepatitis C virus core protein enhances NF-κB signal pathway triggering by lymphotoxin-β receptor ligand and tumor necrosis factor alpha. J. Virol. 1999, 73, 1672–1681. [Google Scholar] [PubMed]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Rendtlew Danielsen, J.M.; Yang, Y.G.; Qi, Y. A role for small RNAs in DNA double-strand break repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Misteli, T. Non-coding RNAs in DNA damage and repair. FEBS Lett. 2013, 587, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Choi, Y.E.; Brault, M.E. Charity begins at home: Non-coding RNA functions in DNA repair. Nat Rev Mol Cell Biol 2013, 14, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Khurana, S.; Kubben, N.; Abdelmohsen, K.; Oberdoerffer, P.; Gorospe, M.; Misteli, T. A BRCA1-interacting lncRNA regulates homologous recombination. EMBO Rep. 2015, 16, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Weiden, M.D.; Ginsberg, H.S. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 1994, 91, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.T.; Schwartz, R.A.; Stracker, T.H.; Lilley, C.E.; Lee, D.V.; Weitzman, M.D. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22, 6610–6620. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Lee, D.V.; Carson, C.T.; Araujo, F.D.; Ornelles, D.A.; Weitzman, M.D. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 2005, 79, 6664–6673. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Bruton, R.K.; Dirlik, O.; Stewart, G.S.; Taylor, A.M.; Dobner, T.; Grand, R.J.; Turnell, A.S. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J. Virol. 2008, 82, 7640–7652. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dodson, G.E.; Shaikh, S.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; You, J.; Benjamin, T.L. Induction and utilization of an ATM signaling pathway by polyomavirus. J. Virol. 2005, 79, 13007–13017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Madden-Fuentes, R.J.; Lou, B.X.; Pipas, J.M.; Gerhardt, J.; Rigell, C.J.; Fanning, E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 2008, 82, 5316–5328. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.H.; Kasper, J.S.; Arai, T.; DeCaprio, J.A. Cul7/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J. Virol. 2004, 78, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Liu, M.T.; Chang, Y.T.; Wu, C.C.; Hu, C.Y.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses DNA repair through the PI3K/Akt/FOXO3a pathway in human epithelial cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.T.; Chen, Y.R.; Chen, S.C.; Hu, C.Y.; Lin, C.S.; Chang, Y.T.; Wang, W.B.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004, 23, 2531–2539. [Google Scholar] [PubMed]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, T.; Verma, S.C.; Lan, K.; Murakami, M.; Robertson, E.S. The ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J. Virol. 2007, 81, 6718–6730. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar] [PubMed]
- Chaurushiya, M.S.; Weitzman, M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair 2009, 8, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Changing the ubiquitin landscape during viral manipulation of the DNA damage response. FEBS Lett. 2011, 585, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Xiaofei, E.; Kowalik, T.F. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 2014, 6, 2155–2185. [Google Scholar] [PubMed]
- Luo, Y.; Qiu, J. Parvovirus infection-induced DNA damage response. Future Virol. 2013, 8, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. “Liver let die”: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.J.; Semmes, O.J. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005, 24, 5986–5995. [Google Scholar] [CrossRef] [PubMed]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005, 12, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Paracha, U.Z.; Fatima, K.; Alqahtani, M.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Qadri, I. Oxidative stress and hepatitis C virus. Virol. J. 2013. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Zimmerman, E.S.; Planelles, V.; Chen, J. Activation of the ATR pathway by human immunodeficiency virus type 1 Vpr involves its direct binding to chromatin in vivo. J. Virol. 2005, 79, 15443–15451. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Shimura, M.; Nakai-Murakami, C.; Tokunaga, K.; Takizawa, Y.; Sata, T.; Kurumizaka, H.; Ishizaka, Y. HIV-1 Vpr induces DNA double-strand breaks. Cancer Res. 2006, 66, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Sherman, M.P.; Blackett, J.L.; Neidleman, J.A.; Kreis, C.; Mundt, P.; Williams, S.A.; Warmerdam, M.; Kahn, J.; Hecht, F.M.; et al. Human immunodeficiency virus type 1 Vpr induces DNA replication stress in vitro and in vivo. J. Virol. 2006, 80, 10407–10418. [Google Scholar] [CrossRef] [PubMed]
- Philpott, S.M.; Buehring, G.C. Defective DNA repair in cells with human T-cell leukemia/bovine leukemia viruses: Role of tax gene. J. Natl. Cancer Inst. 1999, 91, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Bai, X.T.; Shelton, S.; Nicot, C. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS ONE 2012, 7, e42226. [Google Scholar] [CrossRef] [PubMed]
- Chandhasin, C.; Ducu, R.I.; Berkovich, E.; Kastan, M.B.; Marriott, S.J. Human T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage response. J. Virol. 2008, 82, 6952–6961. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Sung, V.M.; Lee, K.J.; Levine, A.M.; Lai, M.M. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 2004, 78, 8835–8843. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 2010, 185, 6985–6998. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Dansako, H.; Abe, K.; Ikeda, M.; Wakita, T.; Kato, N. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J. Virol. 2008, 82, 9639–9646. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Huang, M.; Fang, S.G.; Liu, D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase delta. J. Biol. Chem. 2011, 286, 39546–39559. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Lakshmi, A.N.; Ramana, M.V.; Vijayashree, B.; Ahuja, Y.R.; Sharma, G. Detection of influenza virus induced DNA damage by comet assay. Mutat. Res. 1999, 442, 53–58. [Google Scholar] [CrossRef]
- Clavarino, G.; Claudio, N.; Couderc, T.; Dalet, A.; Judith, D.; Camosseto, V.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; Gatti, E.; et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-beta production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012, 8, e1002708. [Google Scholar] [CrossRef] [PubMed]
- Nargi-Aizenman, J.L.; Simbulan-Rosenthal, C.M.; Kelly, T.A.; Smulson, M.E.; Griffin, D.E. Rapid activation of poly(ADP-ribose) polymerase contributes to Sindbis virus and staurosporine-induced apoptotic cell death. Virology 2002, 293, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, P.; Ruf, M.; Blakqori, G.; Overby, A.K.; Heidemann, M.; Eick, D.; Weber, F. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response-like degradation of transcribing RNA polymerase II. J. Biol. Chem. 2011, 286, 3681–3692. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Austin, D.; Narayanan, A.; Popova, T.; Kainulainen, M.; Bailey, C.; Kashanchi, F.; Weber, F.; Kehn-Hall, K. Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication. J. Biol. Chem. 2012, 287, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Liu, H.J.; Chang, C.D.; Chang, C.I.; Hsu, J.L.; Liao, M.H.; Lee, J.W.; Shih, W.L. Avian reovirus S1133-induced DNA damage signaling and subsequent apoptosis in cultured cells and in chickens. Arch. Virol. 2011, 156, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Guen, V.; Richard, J.; Cohen, E.A.; Berthoux, L. Cell context-dependent involvement of ATR in early stages of retroviral replication. Virology 2010, 396, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Olvera, J.M.; Yoder, K.E.; Mitchell, R.S.; Butler, S.L.; Lieber, M.; Martin, S.L.; Bushman, F.D. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001, 20, 3272–3281. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, L.; Subra, F.; Vaganay, S.; Hervy, M.; Marangoni, E.; Bourhis, J.; Mouscadet, J.F. Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology 2002, 300, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Kanaar, R.; Jackson, S.P.; O’Connor, M.J. Suppression of retroviral infection by the RAD52 DNA repair protein. EMBO J. 2004, 23, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Greger, J.G.; Katz, R.A.; Taganov, K.D.; Wu, X.; Kappes, J.C.; Skalka, A.M. Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol. 2004, 78, 8573–8581. [Google Scholar] [CrossRef] [PubMed]
- Waninger, S.; Kuhen, K.; Hu, X.; Chatterton, J.E.; Wong-Staal, F.; Tang, H. Identification of cellular cofactors for human immunodeficiency virus replication via a ribozyme-based genomics approach. J. Virol. 2004, 78, 12829–12837. [Google Scholar] [CrossRef] [PubMed]
- Baekelandt, V.; Claeys, A.; Cherepanov, P.; De Clercq, E.; de Strooper, B.; Nuttin, B.; Debyser, Z. DNA-Dependent protein kinase is not required for efficient lentivirus integration. J. Virol. 2000, 74, 11278–11285. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Kao, G.; Taganov, K.; Greger, J.G.; Favorova, O.; Merkel, G.; Yen, T.J.; Katz, R.A.; Skalka, A.M. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc. Natl. Acad. Sci. USA 2003, 100, 4778–4783. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Katz, R.A.; Merkel, G.; Hittle, J.C.; Yen, T.J.; Skalka, A.M. Wortmannin potentiates integrase-mediated killing of lymphocytes and reduces the efficiency of stable transduction by retroviruses. Mol. Cell. Biol. 2001, 21, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Swinbank, K.M.; Ahmed, P.S.; Taylor, D.L.; Jackson, S.P.; Smith, G.C.; O’Connor, M.J. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 2005, 7, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Turelli, P.; Masutani, M.; Trono, D. DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type 1 integration. J. Virol. 2005, 79, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Dehart, J.L.; Andersen, J.L.; Zimmerman, E.S.; Ardon, O.; An, D.S.; Blackett, J.; Kim, B.; Planelles, V. The ataxia telangiectasia-mutated and Rad3-related protein is dispensable for retroviral integration. J. Virol. 2005, 79, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, V.J.; Rouleau, M.; Poirier, G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003, 31, 446–454. [Google Scholar] [CrossRef]
- Gaken, J.A.; Tavassoli, M.; Gan, S.U.; Vallian, S.; Giddings, I.; Darling, D.C.; Galea-Lauri, J.; Thomas, M.G.; Abedi, H.; Schreiber, V.; et al. Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J. Virol. 1996, 70, 3992–4000. [Google Scholar] [PubMed]
- Ha, H.C.; Juluri, K.; Zhou, Y.; Leung, S.; Hermankova, M.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. USA 2001, 98, 3364–3368. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, M.; Nukuzuma, S.; Itaya, A.; Tanaka, Y.; Ota, K.; Inada, Y.; Ikuta, K.; Yoshihara, K. Poly(ADP-ribose)polymerase-1 is required for integration of the human immunodeficiency virus type 1 genome near centromeric alphoid DNA in human and murine cells. Biochem. Biophys. Res. Commun. 2005, 334, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Siva, A.C.; Bushman, F. Poly(ADP-ribose) polymerase 1 is not strictly required for infection of murine cells by retroviruses. J. Virol. 2002, 76, 11904–11910. [Google Scholar] [CrossRef] [PubMed]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [PubMed]
- De Murcia, J.M.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.T.; Reyes, D.; Valdes, L.; Saheba, A.; Urias, E.; Mendoza, C.; Fregoso, O.I.; Llano, M. Poly(ADP-ribose) polymerase 1 promotes transcriptional repression of integrated retroviruses. J. Virol. 2013, 87, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, E.; Rossi, O.; Chessa, L.; Frosina, G. Efficient DNA base excision repair in ataxia telangiectasia cells. Eur. J. Biochem. 2000, 267, 6883–6887. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, A.S.; Fishel, R.; Hazuda, D.; Huang, Q.; Xu, M.; Yoder, K.; Zhou, H. siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS ONE 2011, 6, e17612. [Google Scholar] [CrossRef] [PubMed]
- Yoder, K.E.; Espeseth, A.; Wang, X.H.; Fang, Q.; Russo, M.T.; Lloyd, R.S.; Hazuda, D.; Sobol, R.W.; Fishel, R. The base excision repair pathway is required for efficient lentivirus integration. PLoS ONE 2011, 6, e17862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Wang, F.X.; Zhang, H.; Wu, K.J.; Williams, K.J.; Daniel, R. Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair. Virol. J. 2008. [Google Scholar] [CrossRef]
- Tang, H.; Kuhen, K.L.; Wong-Staal, F. Lentivirus replication and regulation. Annu. Rev. Genet. 1999, 33, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.; Rappaport, J. HIV-1 accessory protein Vpr: Relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 2011. [Google Scholar] [CrossRef] [PubMed]
- Jowett, J.B.; Planelles, V.; Poon, B.; Shah, N.P.; Chen, M.L.; Chen, I.S. The human immunodeficiency virus type 1 Vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 1995, 69, 6304–6313. [Google Scholar] [PubMed]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [PubMed]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The human immunodeficiency virus type 1 Vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [PubMed]
- Re, F.; Braaten, D.; Franke, E.K.; Luban, J. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 1995, 69, 6859–6864. [Google Scholar] [PubMed]
- Stewart, S.A.; Poon, B.; Jowett, J.B.; Chen, I.S. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 1997, 71, 5579–5592. [Google Scholar] [PubMed]
- Bartz, S.R.; Rogel, M.E.; Emerman, M. Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J. Virol. 1996, 70, 2324–2331. [Google Scholar] [PubMed]
- Shostak, L.D.; Ludlow, J.; Fisk, J.; Pursell, S.; Rimel, B.J.; Nguyen, D.; Rosenblatt, J.D.; Planelles, V. Roles of p53 and caspases in the induction of cell cycle arrest and apoptosis by HIV-1 Vpr. Exp. Cell Res. 1999, 251, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gelbard, H.A.; Roshal, M.; Pursell, S.; Jamieson, B.D.; Planelles, V. Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J. Virol. 2001, 75, 3791–3801. [Google Scholar] [CrossRef] [PubMed]
- Groschel, B.; Bushman, F. Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. J. Virol. 2005, 79, 5695–5704. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Brasey, A.; Lopez-Lastra, M.; Ohlmann, T.; Beerens, N.; Berkhout, B.; Darlix, J.L.; Sonenberg, N. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J. Virol. 2003, 77, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Nakai-Murakami, C.; Shimura, M.; Kinomoto, M.; Takizawa, Y.; Tokunaga, K.; Taguchi, T.; Hoshino, S.; Miyagawa, K.; Sata, T.; Kurumizaka, H.; et al. HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination. Oncogene 2007, 26, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Zimmerman, E.S.; DeHart, J.L.; Murala, S.; Ardon, O.; Blackett, J.; Chen, J.; Planelles, V. ATR and GADD45alpha mediate HIV-1 Vpr-induced apoptosis. Cell Death Differ. 2005, 12, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Roshal, M.; Kim, B.; Zhu, Y.; Nghiem, P.; Planelles, V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J. Biol. Chem. 2003, 278, 25879–25886. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Chen, J.; Andersen, J.L.; Ardon, O.; Dehart, J.L.; Blackett, J.; Choudhary, S.K.; Camerini, D.; Nghiem, P.; Planelles, V. Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol. Cell. Biol. 2004, 24, 9286–9294. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Elder, R.T.; Qin, K.; Park, H.U.; Liang, D.; Zhao, R.Y. Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. J. Biol. Chem. 2007, 282, 7287–7298. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Park, H.U.; Liang, D.; Zhao, R.Y. Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology 2010. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, E.A. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Monteiro-Filho, C.M.; Arganaraz, E.R.; Planelles, V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 2007, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.P.; Richard, J.; Rougeau, N.; Xiao, Y.; Cohen, E.A. HIV-1 Vpr induces the K48-linked polyubiquitination and proteasomal degradation of target cellular proteins to activate ATR and promote G2 arrest. J. Virol. 2010, 84, 3320–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Mukerjee, R.; Fan, S.; del Valle, L.; Michiels, C.; Sweet, T.; Rom, I.; Khalili, K.; Rappaport, J.; Amini, S.; et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J. Biol. Chem. 2009, 284, 11364–11373. [Google Scholar] [CrossRef] [PubMed]
- Stromajer-Racz, T.; Gazdag, Z.; Belagyi, J.; Vagvolgyi, C.; Zhao, R.Y.; Pesti, M. Oxidative stress induced by HIV-1 F34IVpr in Schizosaccharomyces pombe is one of its multiple functions. Exp. Mol. Pathol. 2010, 88, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Zhang, D.; Hwang, D.S.; Kudchodkar, S.; Dayes, N.S.; Desai, B.M.; Malik, A.S.; Yang, J.S.; Chattergoon, M.A.; Maguire, H.C., Jr.; et al. Adenovirus encoding HIV-1 Vpr activates caspase 9 and induces apoptotic cell death in both p53 positive and negative human tumor cell lines. Oncogene 2002, 21, 4613–4625. [Google Scholar] [CrossRef] [PubMed]
- Shimura, M.; Onozuka, Y.; Yamaguchi, T.; Hatake, K.; Takaku, F.; Ishizaka, Y. Micronuclei formation with chromosome breaks and gene amplification caused by Vpr, an accessory gene of human immunodeficiency virus. Cancer Res. 1999, 59, 2259–2264. [Google Scholar] [PubMed]
- Grassmann, R.; Dengler, C.; Muller-Fleckenstein, I.; Fleckenstein, B.; McGuire, K.; Dokhelar, M.C.; Sodroski, J.G.; Haseltine, W.A. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a Herpesvirus saimiri vector. Proc. Natl. Acad. Sci. USA 1989, 86, 3351–3355. [Google Scholar] [CrossRef] [PubMed]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Higuchi, M.; Makokha, G.N.; Matsuki, H.; Yoshita, M.; Tanaka, Y.; Fujii, M. HTLV-1 Tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood 2013, 122, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Khazaie, K.; Schulze-Osthoff, K.; Baeuerle, P.A.; Schirrmacher, V.; Chlichlia, K. Human T cell leukemia virus-I (HTLV-I) Tax-mediated apoptosis in activated T cells requires an enhanced intracellular prooxidant state. J. Immunol. 1998, 161, 3050–3055. [Google Scholar] [PubMed]
- Kinjo, T.; Ham-Terhune, J.; Peloponese, J.M., Jr.; Jeang, K.T. Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J. Virol. 2010, 84, 5431–5437. [Google Scholar] [CrossRef] [PubMed]
- Chaib-Mezrag, H.; Lemacon, D.; Fontaine, H.; Bellon, M.; Bai, X.T.; Drac, M.; Coquelle, A.; Nicot, C. Tax impairs DNA replication forks and increases DNA breaks in specific oncogenic genome regions. Mol. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; Widen, S.G.; Semmes, O.J.; Wilson, S.H. HTLV-I trans-activator protein, tax, is a trans-repressor of the human beta-polymerase gene. Science 1990, 247, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, T.; Lemoine, F.J.; Donehower, L.A.; Marriott, S.J. Activation of WIP1 phosphatase by HTLV-1 Tax mitigates the cellular response to DNA damage. PLoS ONE 2013, 8, e55989. [Google Scholar] [CrossRef] [PubMed]
- Boxus, M.; Twizere, J.C.; Legros, S.; Kettmann, R.; Willems, L. Interaction of HTLV-1 Tax with minichromosome maintenance proteins accelerates the replication timing program. Blood 2012, 119, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Park, H.U.; Jeong, J.H.; Chung, J.H.; Brady, J.N. Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 2004, 23, 4966–4974. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Guo, X.; Durkin, S.S.; Fryrear, K.F.; Ward, M.D.; Semmes, O.J. Human T-cell leukemia virus type 1 Tax oncoprotein prevents DNA damage-induced chromatin egress of hyperphosphorylated Chk2. J. Biol. Chem. 2007, 282, 29431–29440. [Google Scholar] [CrossRef] [PubMed]
- Haoudi, A.; Semmes, O.J. The HTLV-1 tax oncoprotein attenuates DNA damage induced G1 arrest and enhances apoptosis in p53 null cells. Virology 2003, 305, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.S.; Guo, X.; Fryrear, K.A.; Mihaylova, V.T.; Gupta, S.K.; Belgnaoui, S.M.; Haoudi, A.; Kupfer, G.M.; Semmes, O.J. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 2008, 283, 36311–36320. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Tyrrell, D.L. The cell biology of hepatitis C virus. Microbes Infect. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.D. Hepatitis C virus: Molecular biology & current therapeutic options. Indian J. Med. Res. 2010, 131, 17–34. [Google Scholar] [PubMed]
- Van Pelt, J.F.; Severi, T.; Crabbe, T.; Eetveldt, A.V.; Verslype, C.; Roskams, T.; Fevery, J. Expression of hepatitis C virus core protein impairs DNA repair in human hepatoma cells. Cancer Lett. 2004, 209, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.R.; Sabry, G.M.; Bahnassy, A.A.; Shalaby, K.A.; Abdel-Wahabh, S.A.; Zakaria, S. Mismatch repair genes (hMLH1, hPMS1, hPMS2, GTBP/hMSH6, hMSH2) in the pathogenesis of hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Downregulation of Gadd45beta expression by hepatitis C virus leads to defective cell cycle arrest. Cancer Res. 2010, 70, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Bittar, C.; Shrivastava, S.; Bhanja Chowdhury, J.; Rahal, P.; Ray, R.B. Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS ONE 2013, 8, e62581. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.; Jackwood, M.; Jones, R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [PubMed]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007, 365, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Khadijah, S.; Fang, S.; Wang, L.; Tay, F.P.; Liu, D.X. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010, 84, 8571–8583. [Google Scholar] [CrossRef] [PubMed]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar] [PubMed]
- Schrauwen, E.J.; Fouchier, R.A. Host adaptation and transmission of influenza A viruses in mammals. Emerg. Microbes Infect. 2014, 3, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, M.; Ray, A.; Rawall, S.; Chandna, S.; Kumar, B.; Vijayan, V.K. Detection of influenza virus induced ultrastructural changes and DNA damage. Indian J. Virol. 2010, 21, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.C.; Hong, J.R. RNA viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014, 2014, 467452. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Lobrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7719. [Google Scholar] [CrossRef] [PubMed]
- Col, E.; Caron, C.; Chable-Bessia, C.; Legube, G.; Gazzeri, S.; Komatsu, Y.; Yoshida, M.; Benkirane, M.; Trouche, D.; Khochbin, S. HIV-1 Tat targets Tip60 to impair the apoptotic cell response to genotoxic stresses. EMBO J. 2005, 24, 2634–2645. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, Y.C.; Xu, Q.Z.; Wang, H.P.; Bai, B.; Sui, J.L.; Zhou, P.K. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Zhang, H.; Yang, T.Y.; Ying, T.Y.; Yang, P.X.; Liu, X.D.; Tang, S.J.; Zhou, P.K. Interaction between HIV-1 Tat and DNA-PKcs modulates HIV transcription and class switch recombination. Int. J. Biol. Sci. 2014, 10, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA Damage Response by RNA Viruses. Biomolecules 2016, 6, 2. https://doi.org/10.3390/biom6010002
Ryan EL, Hollingworth R, Grand RJ. Activation of the DNA Damage Response by RNA Viruses. Biomolecules. 2016; 6(1):2. https://doi.org/10.3390/biom6010002
Chicago/Turabian StyleRyan, Ellis L., Robert Hollingworth, and Roger J. Grand. 2016. "Activation of the DNA Damage Response by RNA Viruses" Biomolecules 6, no. 1: 2. https://doi.org/10.3390/biom6010002
APA StyleRyan, E. L., Hollingworth, R., & Grand, R. J. (2016). Activation of the DNA Damage Response by RNA Viruses. Biomolecules, 6(1), 2. https://doi.org/10.3390/biom6010002