
Moderate (2%, v/v) Ethanol Feeding Alters Hepatic Wound Healing after Acute Carbon Tetrachloride Exposure in Mice

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
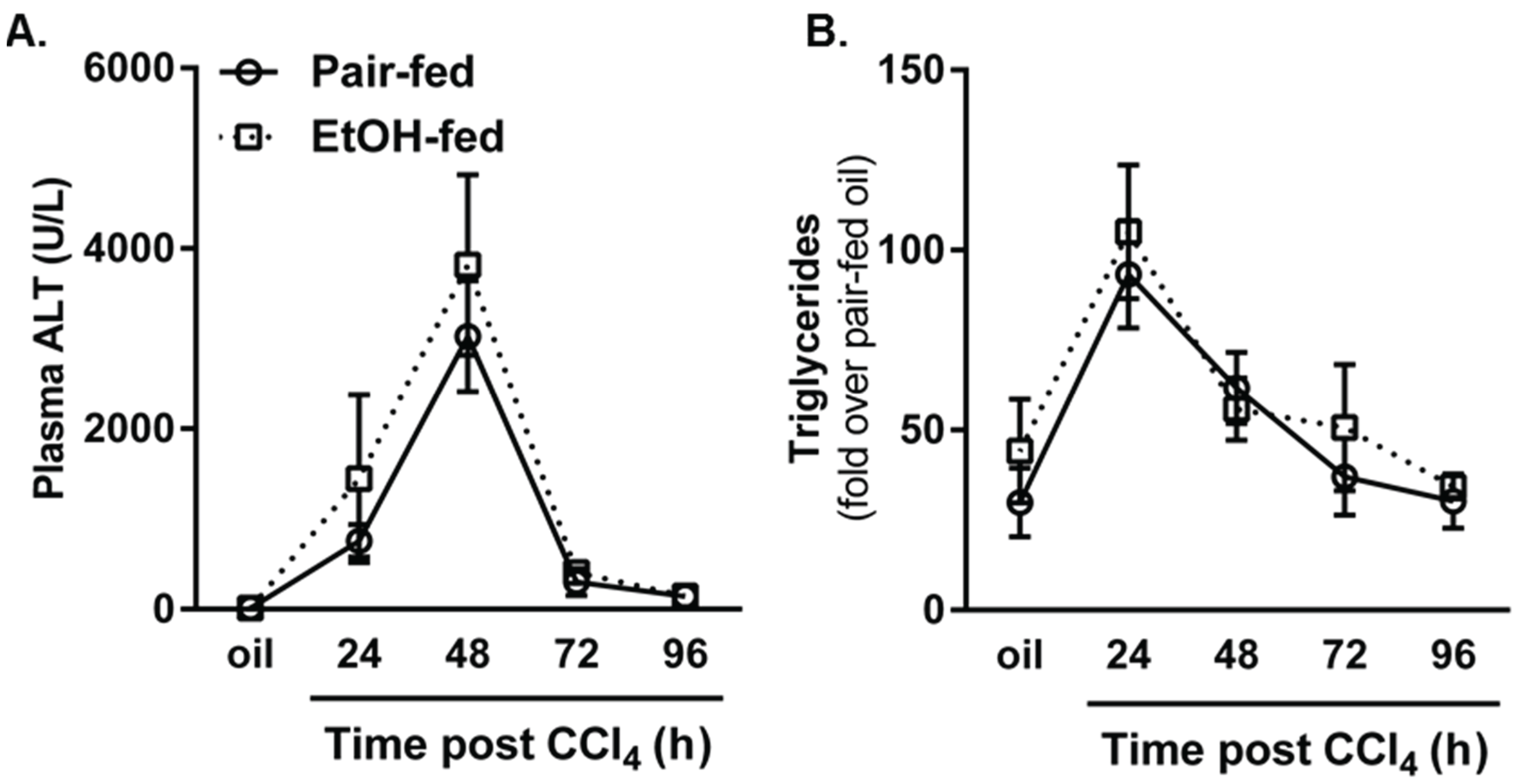
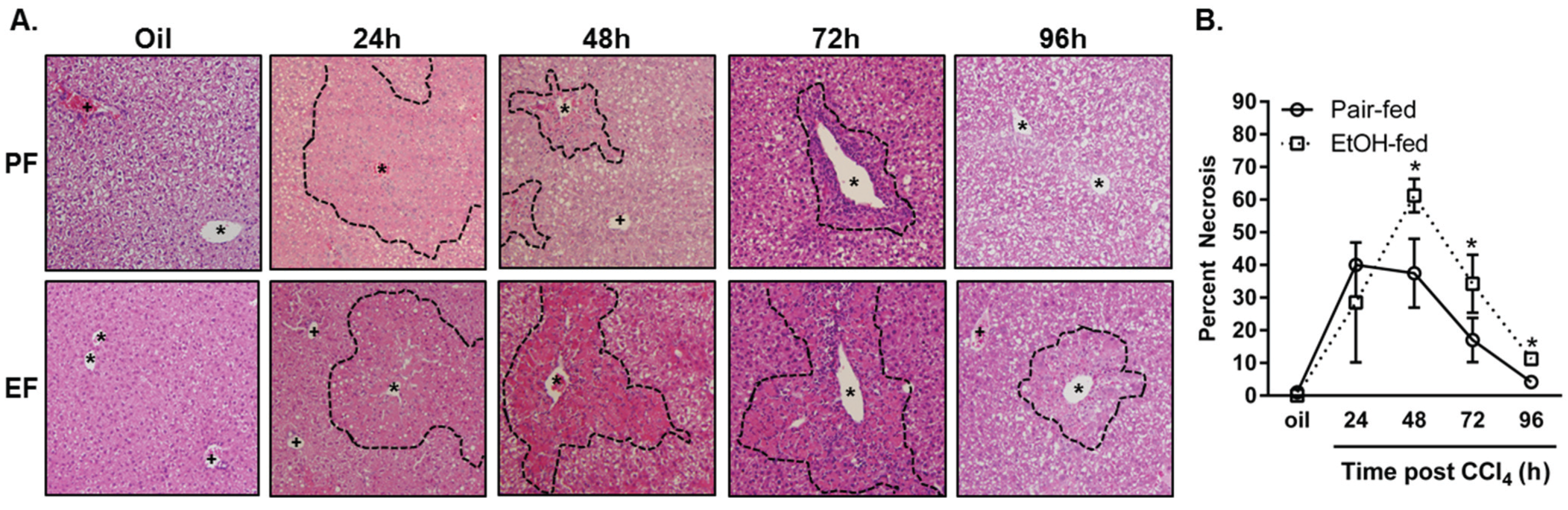
2.1. The Effects of Moderate (2% v/v) Ethanol on Hepatic Cytochrome P450 2E1 (CYP2E1), Hepatic Injury, Steatosis and Removal of Necrotic Tissue after Acute Carbon Tetrachloride (CCl4) Exposure




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAIR-FED | EtOH-FED | |||||||
|---|---|---|---|---|---|---|---|---|
| Exp. Group | Initial BW (g) | Final BW (g) | Liver Weight (g) | Liver to Body Weight Ratio (%) | Initial BW (g) | Final BW (g) | Liver Weight (g) | Liver to Body Weight Ratio (%) |
| Oil | 20.0 (0.9) | 21.2 (1.0) | 0.971 (0.052) | 4.6 (0.250) | 19.1 (0.4) | 20.9 (0.6) | 0.949 (0.066) | 4.5 (0.214) |
| 24 h | 18.8 (0.5) | 21.1 (1.1) | 0.978 (0.056) | 4.6 (0.100) | 17.3 (0.7) | 18.9 (0.6) | 0.964 (0.054) | 5.1 (0.212) * |
| 48 h | 17.7 (0.7) | 19.4 (0.8) | 0.936 (0.031) | 4.7 (0.048) | 18.8 (0.9) | 19.6 (1.0) | 0.988 (0.029) | 5.1 (0.175) * |
| 72 h | 19.7 (0.6) | 20.7 (0.9) | 1.001 (0.068) | 4.8 (0.151) | 19.8 (0.5) | 20.7 (0.8) | 1.081 (0.028) | 5.3 (0.132) * |
| 96 h | 21.4 (0.3) | 21.2 (0.7) | 1.295 (0.038) | 4.9 (0.155) | 21.0 (0.2) | 20.9 (0.2) | 1.115 (0.076) | 5.3 (0.312) |
2.2. Markers of Inflammation and Hepatocyte Apoptosis after Acute CCl4 Exposure: Modulation by Moderate Ethanol
2.2.1. TNFα Production and Hepatic Macrophages
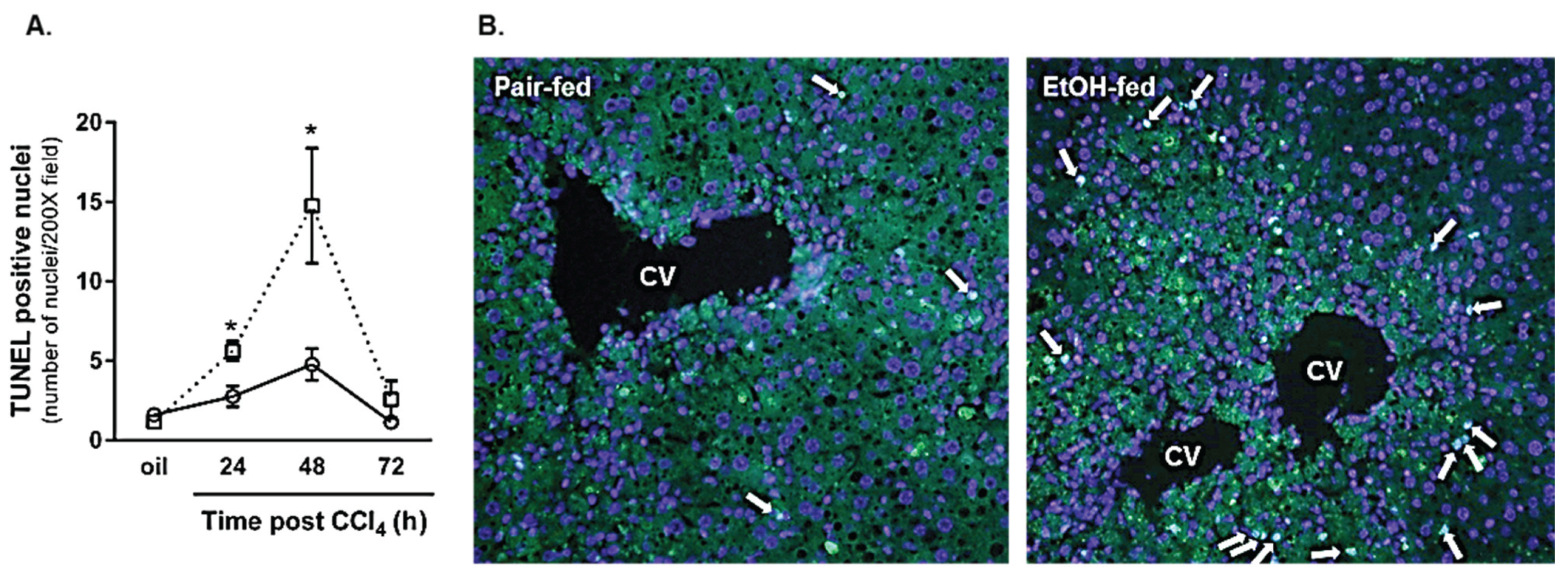
2.2.2. Hepatocyte Apoptosis


2.3. Impact of Moderate Ethanol Feeding to Mice on Liver Regeneration after Acute CCl4
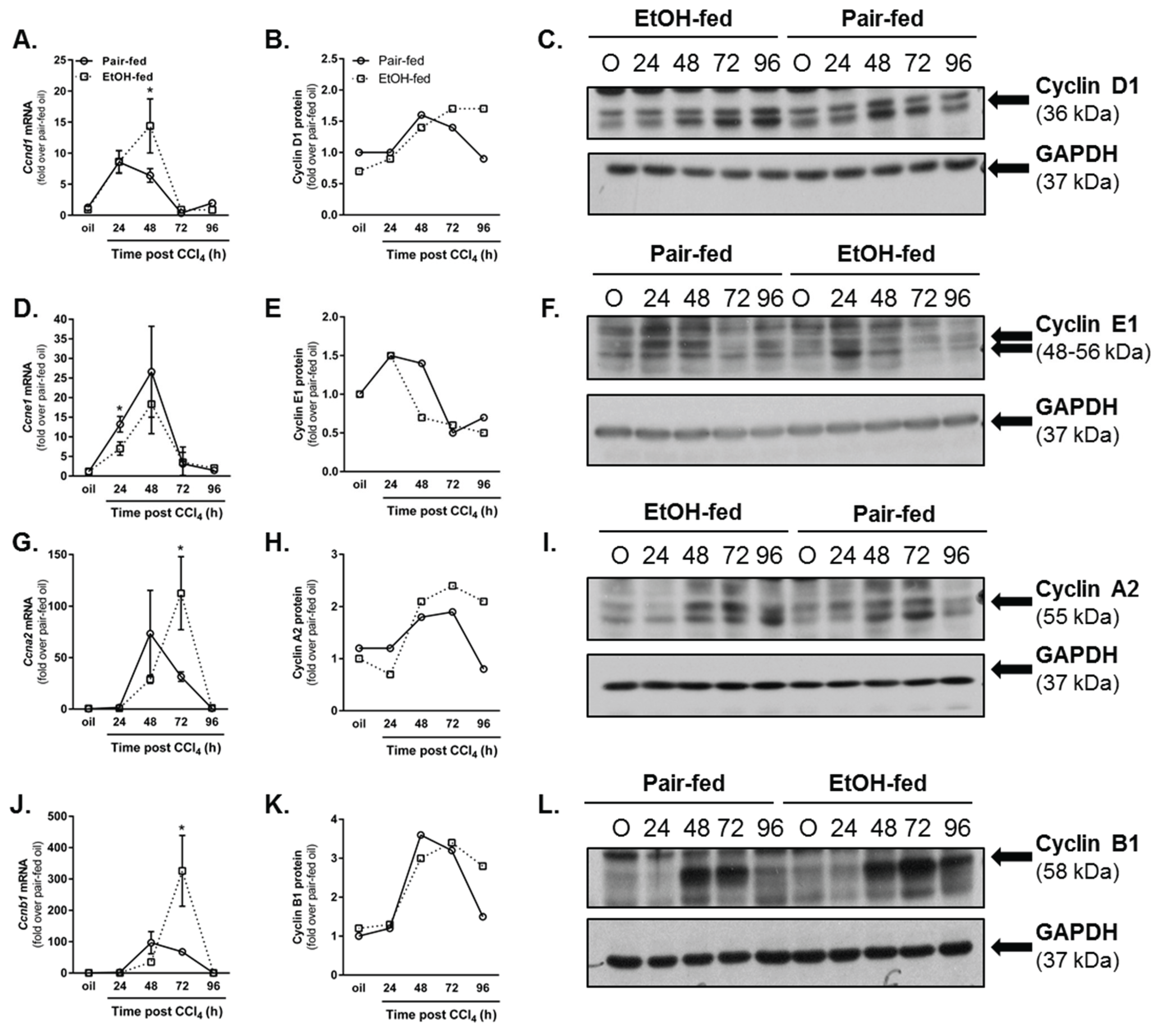
2.3.1. Dynamics of Hepatic Cyclin Content

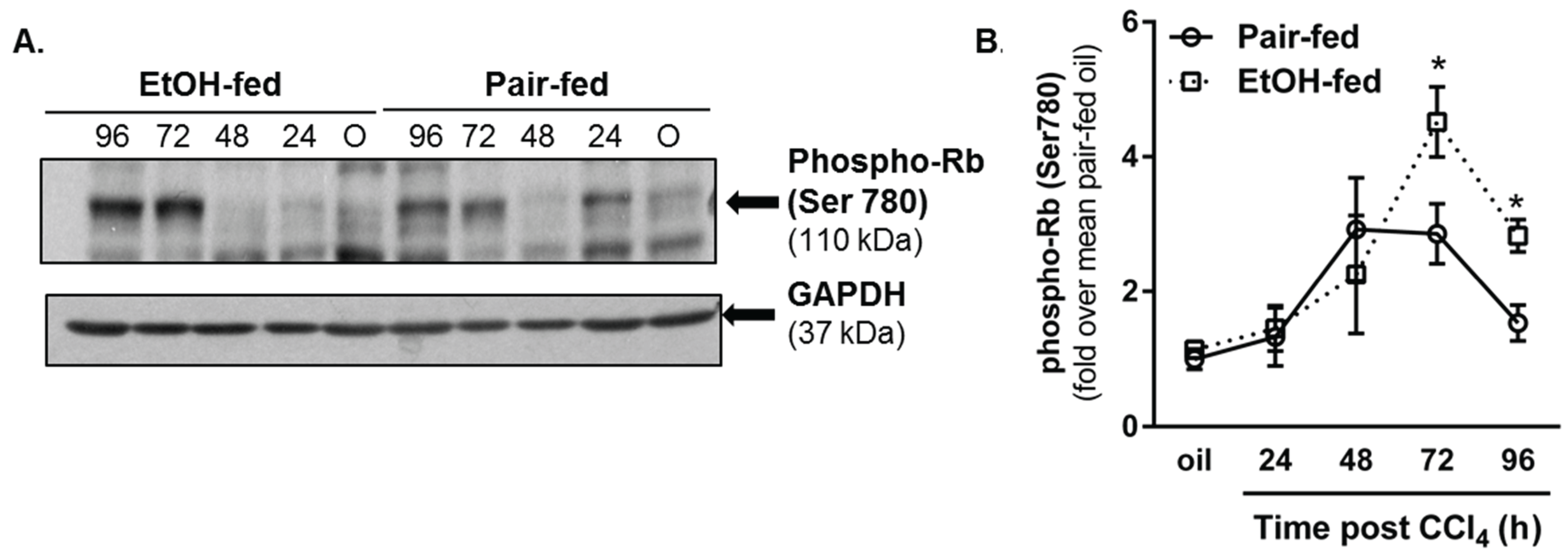
2.3.2. Retinoblastoma Phosphorylation

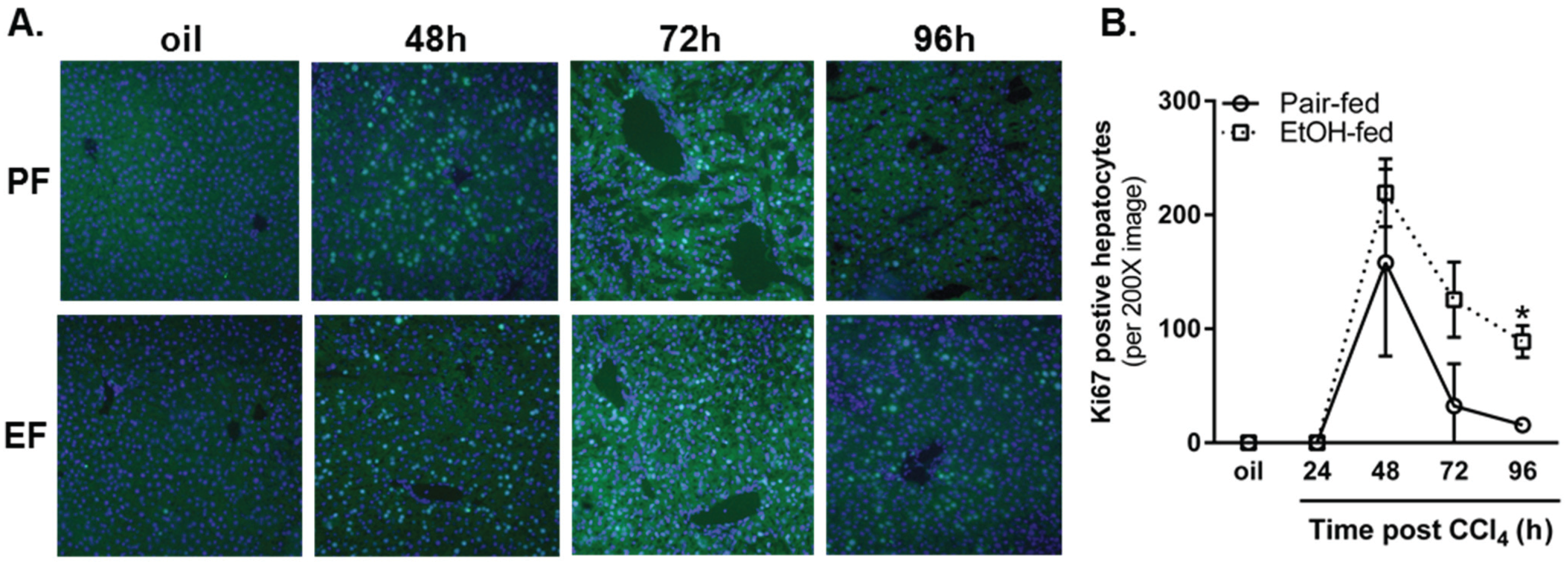
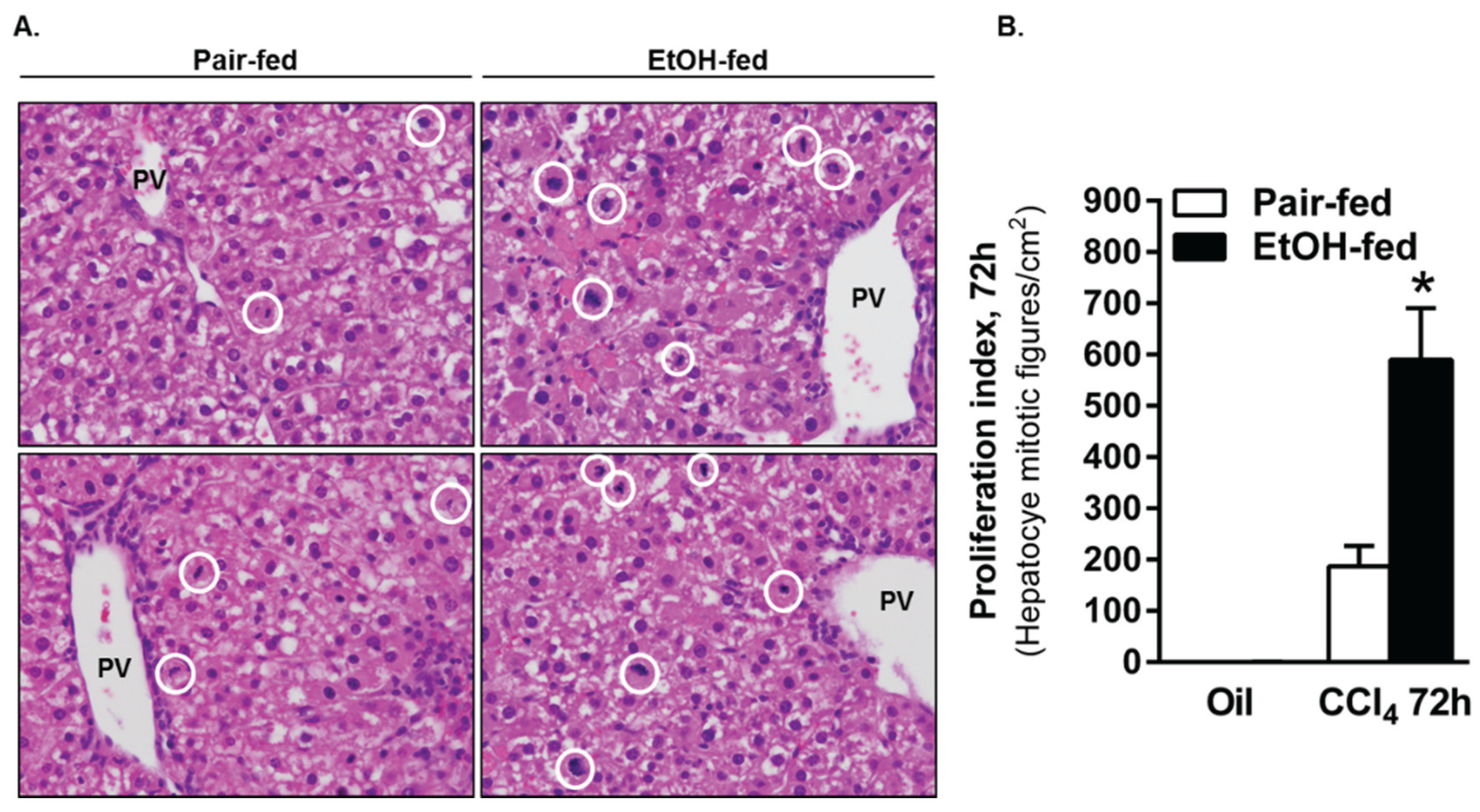
2.3.3. Ki67 Staining and Mitotic Index


2.3.4. Role of Ethanol on Liver Regeneration
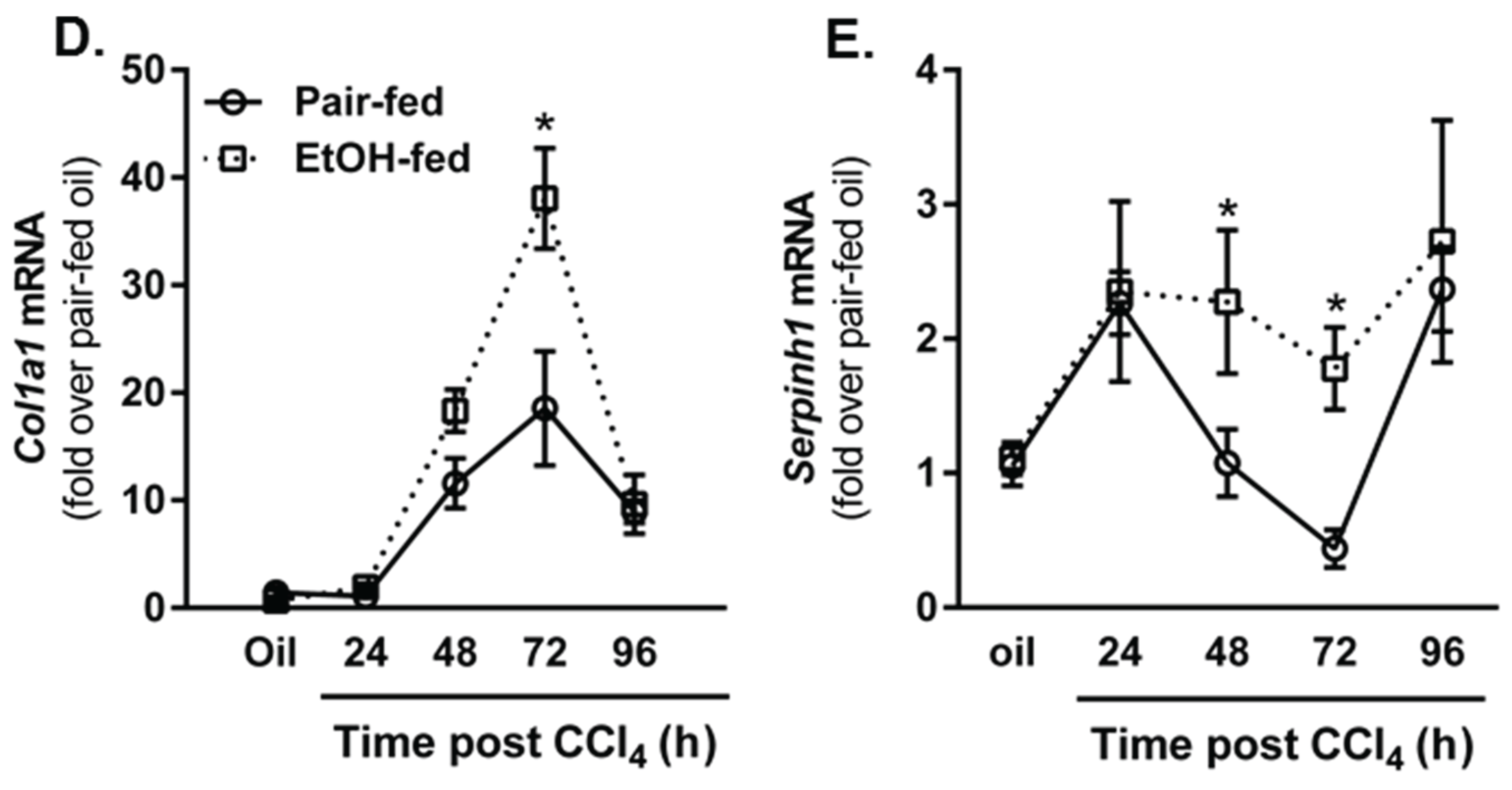
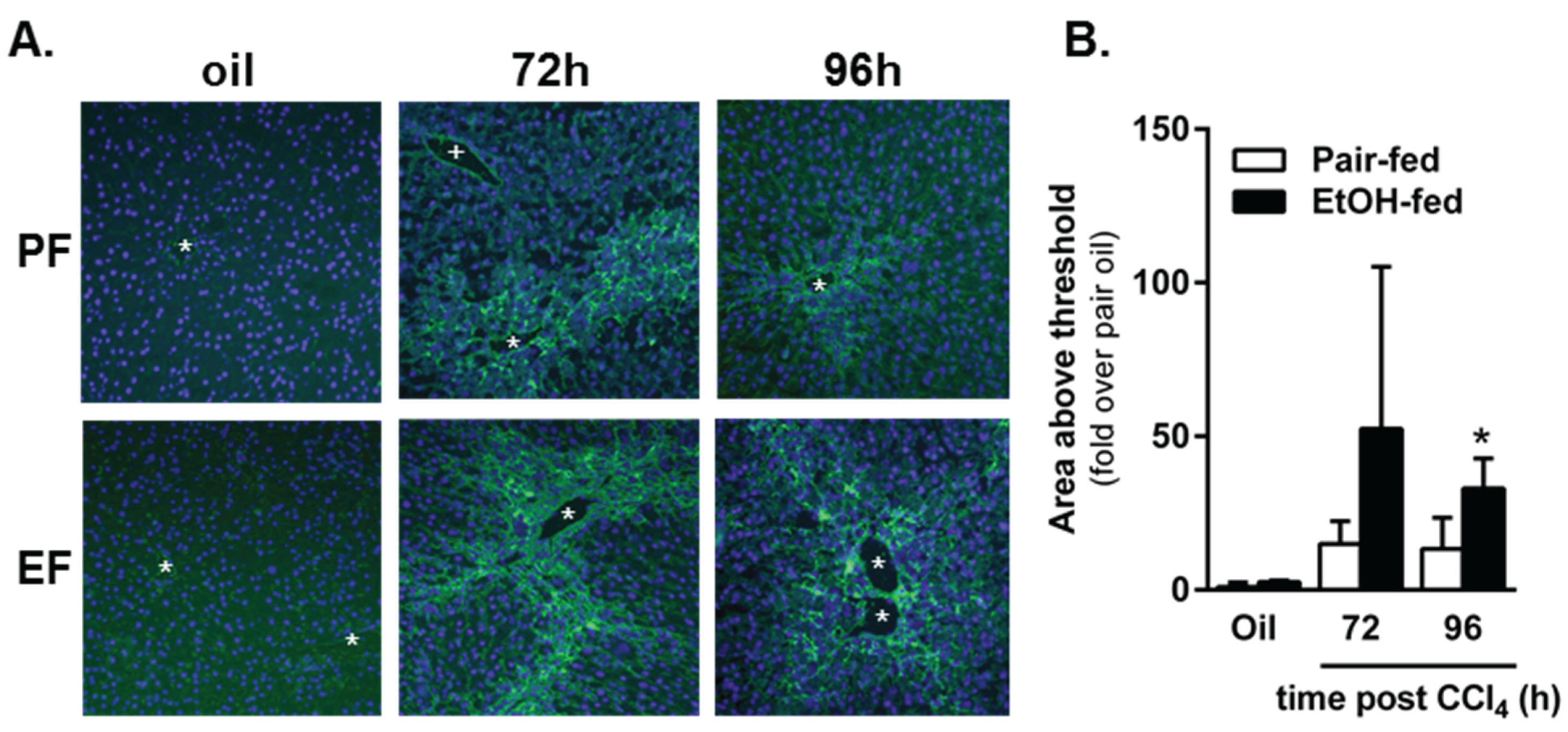
2.4. Impact of Moderate Ethanol Feeding on Hepatic Stellate Cell Activation and Extracellular Matrix (ECM) Remodeling after Acute CCl4 Exposure
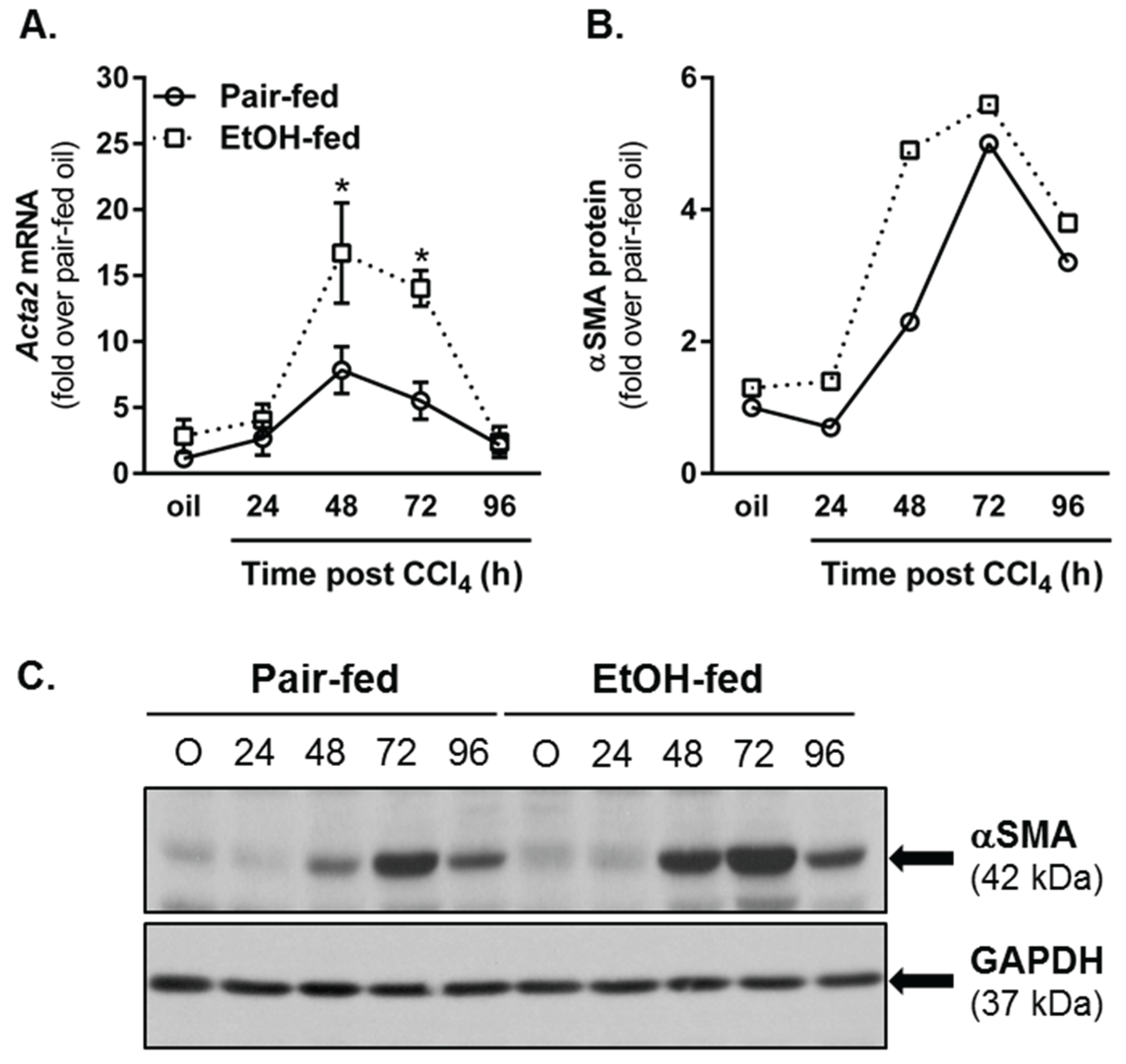
2.4.1. Hepatic Stellate Cell (HSC) Activation


2.4.2. Matrix Remodeling

3. Experimental Section
3.1. Reagents
3.2. Animal Care
3.3. Ethanol Feeding
3.4. Carbon Tetrachloride Exposure, Tissue Collection and Storage
3.5. CYP2E1 Activity Assay
3.6. Liver Injury and Steatosis Determination
3.7. Histopathologic Analysis
3.8. RNA Isolation, cDNA Synthesis and Real-Time Polymerase Chain Reaction (PCR)
| Gene Name | Sequence Source | Forward Primer | Reverse Primer |
|---|---|---|---|
| Tnf | [13] | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Emr1 | PrimerBank: 183583543b1 | CTGCACCTGTAAACGAGGCTT | TTGAAAGTTGGTTTGTCCATTGC |
| Ly6c | PrimerBank: 26353880a1 | GCAGTGCTACGAGTGCTATGG | ACTGACGGGTCTTTAGTTTCCTT |
| Ccnd1 | [52] | CAGAAGTGCGAAGAGGAGGTC | TCATCTTAGAGGCCACGAACAT |
| Ccne1 | PrimerBank: 6671698a1 | GTGGCTCCGACCTTTCAGTC | CACAGTCTTGTCAATCTTGGCA |
| Ccna2 | PrimerBank: 161353443c2 | GCCTTCACCATTCATGTGGAT | TTGCTGCGGGTAAAGAGACAG |
| Ccnb1 | PrimerBank: 28195398a1 | AAGGTGCCTGTGTGTGAACC | GTCAGCCCCATCATCTGCG |
| Acta2 | PrimerBank: 31982518b1 | CCCAGACATCAGGGAGTAATGG | TCTATCGGATACTTCAGCGTCA |
| Col1a1 | [56] | CAAGAACAGCAACGAGTACCG | GTCACTGGTCAACTCCAGCAC |
| Serpinh1 | PrimerBank: 6753304a1 | GCCGAGGTGAAGAAACCCC | CATCGCCTGATATAGGCTGAAG |
3.9. TNFα Enzyme-Linked Immunosorbent (ELISA) Assay
3.10. Terminal Deoxynucleotidyl Transferase-Mediate dUTP Nick End Labeling (TUNEL) Assay Procedure, Image Acquisition and Data Collection
3.11. Immunoblotting
3.12. Ki67 Immunofluorescence Assay, Image Acquisition and Data Collection
3.13. Mitotic Figure Quantification
3.14. In Situ Zymography, Image Acquisition and Data Collection
3.15. Statistics
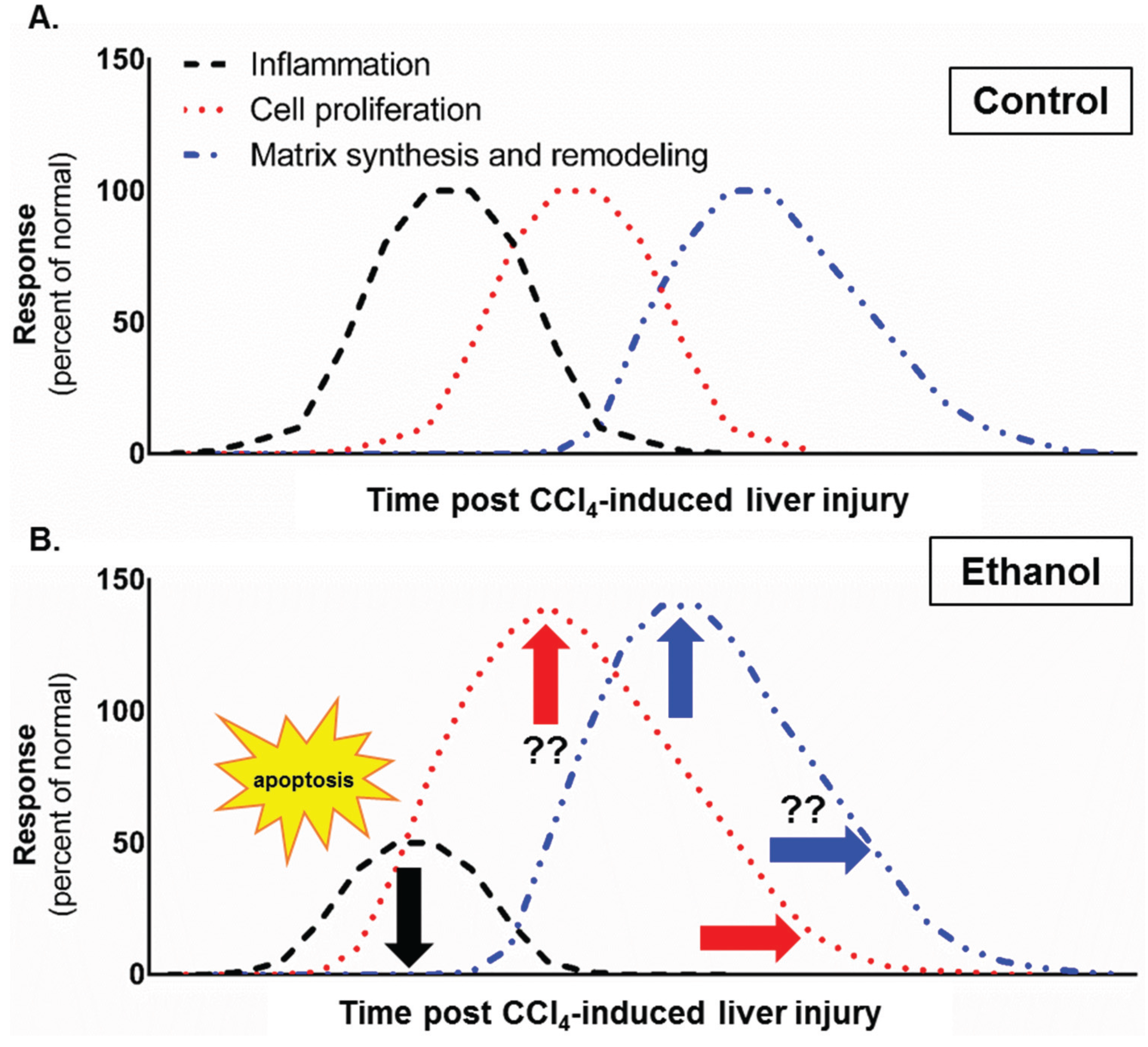
4. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bataller, R.; Rombouts, K.; Altamirano, J.; Marra, F. Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Prac. Res. Clin. Gastroenterol. 2011, 25, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.; Xu, M.; Wang, H.; Bertola, A.; Gao, B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: Pathophysiology, translational relevance, and challenges. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G819–G823. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-γ contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.J.; Roychowdhury, S.; Bush, K.; McMullen, M.R.; Pisano, S.; Niese, K.; Olman, M.A.; Pritchard, M.T.; Nagy, L.E. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS ONE 2013, 8, e69114. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; Chiang, D.J.; McMullen, M.R.; Nagy, L.E. Moderate, chronic ethanol feeding exacerbates carbon-tetrachloride-induced hepatic fibrosis via hepatocyte-specific hypoxia inducible factor 1α. Pharmacol. Res. Perspect 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; Chiang, D.J.; Mandal, P.; McMullen, M.R.; Liu, X.; Cohen, J.I.; Pollard, J.; Feldstein, A.E.; Nagy, L.E. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl4-induced fibrosis but not steatosis or inflammation. Alcohol. Clin. Exp. Res. 2012, 36, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Teoh, N.; Leclercq, I.; Pena, A.D.; Farrell, G. Low-dose TNF-α protects against hepatic ischemia-reperfusion injury in mice: Implications for preconditioning. Hepatology 2003, 37, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Teoh, N.C.; Farrell, G.C. Hepatic ischemia reperfusion injury: Pathogenic mechanisms and basis for hepatoprotection. J. Gastroenterol. Hepatol. 2003, 18, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, M.T.; Cohen, J.I.; Roychowdhury, S.; Pratt, B.T.; Nagy, L.E. Early growth response-1 attenuates liver injury and promotes hepatoprotection after carbon tetrachloride exposure in mice. J. Hepatol. 2010, 53, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Webber, E.M.; Bruix, J.; Pierce, R.H.; Fausto, N. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology 1998, 28, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Fausto, N. Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am. J. Pathol. 1998, 152, 1577–1589. [Google Scholar] [PubMed]
- Yamada, Y.; Kirillova, I.; Peschon, J.J.; Fausto, N. Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Webber, E.M.; Kirillova, I.; Peschon, J.J.; Fausto, N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: Requirement for type 1 but not type 2 receptor. Hepatology 1998, 28, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.W.; Chan, W.Y.; Lee, S.S. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol. Appl. Pharmacol. 1998, 153, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.; Boll, M.; Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Lindros, K.O. Zonation of cytochrome p450 expression, drug metabolism and toxicity in liver. Gen. Pharmacol. 1997, 28, 191–196. [Google Scholar] [CrossRef]
- Mehendale, H.M. Tissue repair: An important determinant of final outcome of toxicant-induced injury. Toxicol. Pathol. 2005, 33, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Mehendale, H.M. Once initiated, how does toxic tissue injury expand? Trends Pharmacol. Sci. 2012, 33, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Aisaki, K.; Ikawa, Y.; Wake, K. Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am. J. Pathol. 1998, 153, 515–525. [Google Scholar] [CrossRef]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radical Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Kenney, F.T.; Pomato, N.; Lee, K.L. Mechanisms in intracellular turnover of stable and labile enzymes. In Protein Turnover and Lysosome Function; Segal, H.L., Doyle, D.J., Eds.; Academic Press, Inc: New York, NY, USA, 1978; pp. 197–213. [Google Scholar]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Prosser, C.C.; Yen, R.D.; Wu, J. Molecular therapy for hepatic injury and fibrosis: Where are we? World J. Gastroenterol. 2006, 12, 509–515. [Google Scholar] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; McMullen, M.R.; Pritchard, M.T.; Hise, A.G.; van Rooijen, N.; Medof, M.E.; Stavitsky, A.B.; Nagy, L.E. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology 2009, 49, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Iredale, J.P. Macrophages: Central regulators of hepatic fibrogenesis and fibrosis resolution. J. Hepatol. 2012, 56, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Viebahn, C.S.; Yeoh, G.C. What fires prometheus? The link between inflammation and regeneration following chronic liver injury. Int. J. Biochem. Cell Biol. 2008, 40, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Strey, C.W.; Markiewski, M.; Mastellos, D.; Tudoran, R.; Spruce, L.A.; Greenbaum, L.E.; Lambris, J.D. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J. Exp. Med. 2003, 198, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.P.; Liljequist, B.L.; Bartizal, K.F. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology 1990, 11, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Moroy, T.; Geisen, C. Cyclin E. Int. J. Biochem. Cell Biol. 2004, 36, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Pines, J.; Hunter, T. Cyclins A and B1 in the human cell cycle. Ciba Found. Symp. 1992, 170, 187–196. [Google Scholar] [PubMed]
- Desdouets, C.; Sobczak-Thepot, J.; Murphy, M.; Brechot, C. Cyclin A: Function and expression during cell proliferation. Prog. Cell Cycle Res. 1995, 1, 115–123. [Google Scholar] [PubMed]
- Kishimoto, T.; Okumura, E. In vivo regulation of the entry into M-phase: Initial activation and nuclear translocation of cyclin B/Cdc2. Prog. Cell Cycle Res. 1997, 3, 241–249. [Google Scholar] [PubMed]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Beier, J.I.; Baldauf, K.J.; Jokinen, J.D.; Zhong, H.; Arteel, G.E. Acute ethanol preexposure promotes liver regeneration after partial hepatectomy in mice by activating ALDH2. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G37–G47. [Google Scholar] [CrossRef] [PubMed]
- Apte, U.M.; McRee, R.; Ramaiah, S.K. Hepatocyte proliferation is the possible mechanism for the transient decrease in liver injury during steatosis stage of alcoholic liver disease. Toxicol. Pathol. 2004, 32, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wands, J.R.; Carter, E.A.; Bucher, N.L.; Isselbacher, K.J. Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology 1979, 77, 528–531. [Google Scholar] [PubMed]
- Diehl, A.M.; Wells, M.; Brown, N.D.; Thorgeirsson, S.S.; Steer, C.J. Effect of ethanol on polyamine synthesis during liver regeneration in rats. J. Clin. Invest. 1990, 85, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Koteish, A.; Yang, S.; Lin, H.; Huang, J.; Diehl, A.M. Ethanol induces redox-sensitive cell-cycle inhibitors and inhibits liver regeneration after partial hepatectomy. Alcohol. Clin. Exp. Res. 2002, 26, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.E.; Broadhurst, K.A.; Mathahs, M.M.; Brunt, E.M.; Schmidt, W.N. Expression of HSP47, a collagen-specific chaperone, in normal and diseased human liver. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, M.T.; Malinak, R.N.; Nagy, L.E. Early growth response (EGR)-1 is required for timely cell-cycle entry and progression in hepatocytes after acute carbon tetrachloride exposure in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1124–G1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seed, B. A PCR primer bank for quantitative gene expression analysis. Nucl. Acids Res. 2003. [Google Scholar] [CrossRef]
- Spandidos, A.; Wang, X.; Wang, H.; Dragnev, S.; Thurber, T.; Seed, B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 2008. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Seed, B. Primerbank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010, 38, D792–D799. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, M.T.; Nagy, L.E. Hepatic fibrosis is enhanced and accompanied by robust oval cell activation after chronic carbon tetrachloride administration to Egr-1-deficient mice. Am. J. Pathol. 2010, 176, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- Nova, E.; Baccan, G.C.; Veses, A.; Zapatera, B.; Marcos, A. Potential health benefits of moderate alcohol consumption: Current perspectives in research. Proc. Nutr. Soc. 2012, 71, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Neafsey, E.J.; Mukamal, K.J.; Gray, M.O.; Parks, D.A.; Das, D.K.; Korthuis, R.J. Alcohol in moderation, cardioprotection, and neuroprotection: Epidemiological considerations and mechanistic studies. Alcohol. Clin. Exp. Res. 2009, 33, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Baldwin, L.A. Ethanol and hormesis. Crit. Rev. Toxicol. 2003, 33, 407–424. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshpande, K.T.; Liu, S.; McCracken, J.M.; Jiang, L.; Gaw, T.E.; Kaydo, L.N.; Richard, Z.C.; O’Neil, M.F.; Pritchard, M.T. Moderate (2%, v/v) Ethanol Feeding Alters Hepatic Wound Healing after Acute Carbon Tetrachloride Exposure in Mice. Biomolecules 2016, 6, 5. https://doi.org/10.3390/biom6010005
Deshpande KT, Liu S, McCracken JM, Jiang L, Gaw TE, Kaydo LN, Richard ZC, O’Neil MF, Pritchard MT. Moderate (2%, v/v) Ethanol Feeding Alters Hepatic Wound Healing after Acute Carbon Tetrachloride Exposure in Mice. Biomolecules. 2016; 6(1):5. https://doi.org/10.3390/biom6010005
Chicago/Turabian StyleDeshpande, Krutika T., Shinlan Liu, Jennifer M. McCracken, Lu Jiang, Ta Ehpaw Gaw, Lindsey N. Kaydo, Zachary C. Richard, Maura F. O’Neil, and Michele T. Pritchard. 2016. "Moderate (2%, v/v) Ethanol Feeding Alters Hepatic Wound Healing after Acute Carbon Tetrachloride Exposure in Mice" Biomolecules 6, no. 1: 5. https://doi.org/10.3390/biom6010005
APA StyleDeshpande, K. T., Liu, S., McCracken, J. M., Jiang, L., Gaw, T. E., Kaydo, L. N., Richard, Z. C., O’Neil, M. F., & Pritchard, M. T. (2016). Moderate (2%, v/v) Ethanol Feeding Alters Hepatic Wound Healing after Acute Carbon Tetrachloride Exposure in Mice. Biomolecules, 6(1), 5. https://doi.org/10.3390/biom6010005